It has been established [2] that the redox properties of the ligand vary with the incorporated metal while in turn the metal redox properties can be controlled by the peripheral substituents of the porphyrin placed at the b and /or meso positions.From this point of view, electron-withdrawing substituents are particularly interesting since they lead to electron-deficient porphyrins which are generally less easily oxidized than the parent compound. Nevertheless, these substituents can affect the conformation of the macrocycle and hence the redox potentials [3]. Although the balanced final effects leading or not to the porphyrin HOMO stabilization are not fully predictable à priori, an enhanced catalyst stability as well as catalytic activity can be reasonably expected from these new porphyrins.
As strong electron-withdrawing substituents with a neat -I character, the perfluoroalkyl groups have received a great attention. Kaesler and LeGoff prepared the first porphyrins bearing eight perfluoroalkyl chains on the pyrrole rings by acid-catalyzed self-condensation of functionalized derivatives of 2,5-dimethyl-3,4-bis(polyfluoroalkyl) pyrroles [4]. A methylene unit was introduced between the perfluoroalkyl chain and the pyrrole ring since trifluoromethyl groups, when directly placed in the 3 and 4-positions, were found to totally deactivate the pyrrole nucleus vs. tetramerization. Some other porphyrins bearing one trifluoromethyl group by pyrrole nucleus were prepared from 3-(trifluoromethyl) pyrroles derivatives [5]. Recently, a meso-tetrakis (perfluoroalkyl)porphyrin has been prepared from an (hydroxymethyl)pyrrole precursor [3a].
Chlorine and bromine, electron-withdrawing groups with an additional +M character, have been attached to the porphyrin ring. For example, iron tetraarylporphyrins with halogenated pyrrole units and protected meso positions were found to be efficient catalysts for the hydroxylation of poorly reactive alkanes [6]. While these halogens were introduced on the pyrrole rings of meso-substituted porphyrins essentially with N-bromo or N-chlorosuccinimide [3b and references therein], the introduction of fluorine was far more problematic.
Although fluorinated porphyrins were expected to have unique physico- and biochemical properties, examples of genuine fluoroporphyrins i.e. bearing fluorine atoms directly linked to the macrocycle remain scarce to date. The first reported one, 2,7,12,17-tetrafluoro-3,8,13,18-tetramethylporphyrin, was synthetized by Ogoshi et al. [7] in 2% yield by tetramerization of the acid 4-fluoro-3-methyl-5-(hydroxymethyl)pyrrole-2-carboxylic. The second reported example in the opened litterature was obtained by substitution of all the pyrrole protons of a Zn metallated meso-tetra(pentafluorophenyl)porphyrin by fluorine, with silver fluoride or cobalt fluoride. Unfortunately, experimental details were lacking as well as the valence of the silver used, probably (II). The perfluorinated porphyrin obtained, as its iron (III), Cl- complex form, was found to catalyze the hydroxylation of benzene into phenol, by hydrogen peroxide at room temperature and atmospheric pressure [6c]. Other examples were focused on meso-fluorination of porphyrins. This position was known to be susceptible to electrophilic halogen substitution [8]. Meso-fluoro derivatives of deuterioporphyrin were obtained in low yields via a Schiemann reaction [9]. More recently, direct fluorination of octaethylporphyrin was performed with caesium fluoroxysulphate, the major product being the 5-fluoroderivative [10]. In our hands, direct fluorination of a meso-substituted porphyrin with diluted fluorine at low temperature was deceptive.
While it appears from the litterature that no (per)chloro or (per)bromoporphyrins have been prepared by classical porphyrins synthesis e.g.the Adler-Longo methodology [11] from 3,4-dichloro or dibromopyrroles derivatives, the success of Onda et al. [7] with a b-fluoropyrrole encouraged us to explore this approach starting from 3,4-difluoropyrrole derivatives.
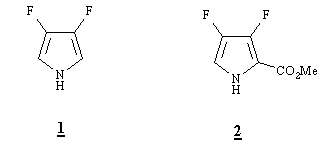
We recently described the synthesis of 3,4-difluoropyrrole (1) from 1-tert-butyl-2-(methoxycarbonyl)aziridine and chlorotrifluoroethylene [12]. We sought that the intermediate ester, 2-(methoxycarbonyl-3,4-difluoropyrrole) (2) could be a synthon of choice to acceed to 2,3,7,8,12,13,17,18-octafluoroporphyrin [OFP].
In a recent study devoted, inter alia, to the synthesis of octaethylporphyrin [OEP] [13], 2-ethoxycarbonyl-3,4-diethylpyrrole was converted in a one-pot procedure to OEP via 3,4-diethylpyrrole generated in situ by fast demethylation with LiCl in wet DMSO at 200°C and subsequent thermal decarboxylation. In the presence of paraformaldehyde, the Lewis acidic Li+ cation catalyzed the formation of porphyrinogen which was oxidized by oxygen to afford OEP in 17% yield. As far better yields (67%) of OEP were obtained from a preformed dipyrromethane diester using the same dealkylation-decarboxylation procedure in the presence of paraformaldehyde and air bubbling, we prepared the fairly stable dipyrromethane 3 in 85% yield from 2 and dimethoxymethane with boron trifluoride diethyl etherate as catalyst. Reaction was complete within 3-4 days at room temperature as monitored by TLC. In the same conditions, 2-(ethoxycarbonyl)-3-(trifluoromethyl)-4-ethylpyrrole [13] was converted into the corresponding dipyrromethane within 35 h. This difference of reactivity could indicate that the deactivating effect of the two fluorine atoms vs. electrophilic substitution at the 5-position is not negligible as compared to that of the powerful electron-withdrawing trifluoromethyl group.
Preliminary experiments of direct demethylation-decarboxylation of the ester 2 with LiCl in DMSO at 200°C showed that this process was rapid after a short initiation time. For example, after injection of a solution of ca.1.7 mmol of 2 in DMSO (1 mL) in a pre-heated stirred suspension of LiCl (10 equiv) in DMSO (8 mL) and water (1 equiv), the theoretical volume of carbon dioxide (ca. 39 mL) was evolved within 10 mn. After quenching over ice, the mixture was extracted with diethyl ether and the combined organic extracts dried. Careful distillation of the ether afforded impure oily 1 which was distilled twice in a short-path distillation apparatus to give crystallized 1 in 23% yield. Analytical data (NMR, MS) were in full accordance with that of an authentic sample [12]. In the same conditions and although a theoretical volume of carbon dioxide was collected, isolation of any tractable product from the reaction of dipyrromethane 3 with LiCl failed. Nevertheless, a preparation of OFP from 3 in the conditions of Tang and Verkade[13] (LiCl, water, and paraformaldehyde in DMSO at 200°C) was attempted. Only a coal was obtained.
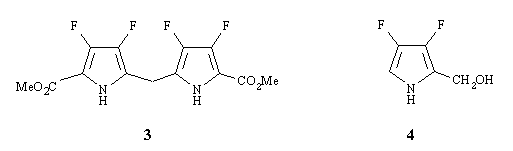
As reported by others [7][13], a 2-(hydroxymethyl)pyrrole is also a synthon of choice. For example, OEP was prepared in 69% yield under acid conditions (p-toluenesulfonic acid, PTSA) from 2-(hydroxymethyl)-3,4-diethylpyrrole with dimethoxymethane as a dehydrating agent [13]. The ester 2 was selectively reduced to 2-(hydroxymethyl)-3,4-difluoropyrrole (4) with lithium-aluminium hydride. The reaction which was performed at 0-5°C in dry THF was complete within ca. 3 h (TLC monitoring). This alcohol proved to be essentially unstable, like its 3,4-diethyl analog [13], decomposing on standing at room temperature within few hours to give a coal. Nevertheless, an OFP synthesis could be attempted when the crude product was used rapidly without further purification. When PTSA was added at room temperature to a mixture of 4 and an excess of dimethoxymethane in dichloromethane, an immediate reaction occured owing to the apparition of an orange colouring fastly turning brown. Examination by mass spectrometry of the crude mixture (an aliquot was stored for ca. 20 h at -18°C) revealed the presence of octafluoroporphyrinogen, the reduced form of OFP (Theoretical mass. /z 460) Found (CI+NH3) m/ 461 (M+1), m/z 477 (M+17). Examination by mass spectrometry of the mixture which was left to react at room temperature for the same time (20 h), indicated the disappearence of octafluoroporphyrinogen without appearence of OFP.
In conclusion, although OFP formation has not been not observed for the moment, the above preliminary results underline the complex reactivity of 3,4-difluoropyrrole derivatives as compared to that of the alkyl analogs.
1 Frank, B. Angew. Chem., Int. Ed. Engl. 1982, 21, 343.
2 Felton, R. H.; Davis, D. G. in The Porphyrins, Dolphin, D., Ed.; Academic Press: New York, 1978; Vol 5, chapters 3 and 4.
3 (a) DiMagno, S. G.; Williams, R. A.; Therien, M. J. J. Org. Chem. 1994, 59, 6943-6948; (b) Ochsenbein, P.; Ayougou, K.; Mandon, D.; Fischer, J.; Weiss, R.; Austin, R. N.; Jayaraj, K.; Gold, A.; Terner, J.; Fajer, J. Angew. Chem. Int. Ed. Engl. 1994, 33, 348-350.
4 Kaesler, R. W.; LeGoff, E. J. Org. Chem. 1982, 47, 5243-5246.
5 (a) Homma, M.; Aoyagi, Y.; Aoyama, Y.; Ogoshi, H. Tetrahedron Lett. 1975, 24, 4343-4346. (b) Toi, H.; Homma, A.; Suzuki, A.; Ogoshi, H. J. Chem. Soc., Chem. Commun. 1985, 1791-1792.
6 (a) Traylor, T. G.; Tsuchiya, S. Inorg. Chem. 1987, 26, 1338-1339; (b) Wijesekera, T. P.; Matsumoto, A.; Dolphin, D.; Lexa, D. Angew. Chem. Int. Ed. Engl. 1990; 29, 1028-1029; (c) Tsuchiya, S.; Seno, M. Chem. Lett. 1989, 263- 266.
7 Onda, H.; Toi, H.; Ayoama, Y.; Ogoshi, H. Tetrahedron Lett. 1985, 26, 4221-4224.
8 (a) Woodward, R. B.; Skavic, V. J. Am. Chem. Soc. 1961, 83, 4676-4678; (b) Bonnett, R.; Gale, I. A. D., Stephenson, G. F. J. Chem. Soc. C 1966, 1600-1604.
9 Baker, E. W.; Billig, M. J. Chem. Ind. 1969, 654.
10 Andrews, L. E.; Bonnnett, R.; Kozyrev, A. N.; Appelman, E. H. J. Chem. Soc., Perkin trans 1 1988, 1735-1738.
11 Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827 and references therein.
12 Leroy, J.; Wakselman, C. Tetrahedron Lett. 1994, 35, 8605-8608.
13 Tang, J.; Verkade, J. G. J. Org. Chem. 1994, 59, 7793-7802.