An Unexpected Tandem Reaction of N-Butadienyl-N-Alkylketene-N,O-Silylacetals

In recent years some spectacular applications of tandem reactions to the synthesis of complex natural products have been reported using either the biomimetic approach5 or tandem reactions specifically designed for their synthetic utility6. Lately the interest in developing new tandem reactions has increased considerably. The systematic search for tandem reactions has been motivated by the recognition of the higher efficiency which can be obtained using tandem reactions instead of a stepwise procedure 7,8. Recently a systematic nomenclature for tandem reactions has been proposed and reviews on tandem reactions have been published.
In connection with our interest in tandem reactions composed of a Diels-Alder reaction and a [3,3]-sigmatropic shift9,10,11 N-butadienyl-N-isopropylketene-N,O-trimethylsilylacetal of propionamide has been synthesised and its reactivity against N-phenylmaleimide and acryloyl chloride have been tested 12,13(figure 1).
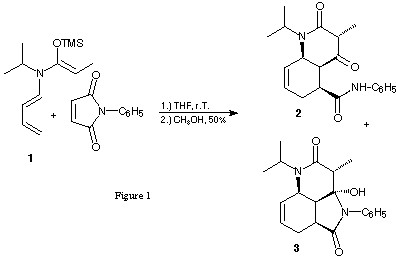
Instead of the planned tandem reaction Diels-Alder reaction/[3,3]-sigmatropic shift an unexpected tandem reaction Diels-Alder reaction/acylation was observed leading to interesting bicyclic and tricyclic products 2,3 and 4 in good yields (see figure 1). The combination of a Diels-Alder reactions with an acylations has not been used very often. Most of the reported cases14,15,16,17 are one-pot reactions starting with the N-acylations of the adequate imines with acid chlorides forming a C-N bond first leading to an N-butadienyl amide, which is itself the starting material for the subsequent Diels-Alder reaction. In contrast to the cases reported in the literature our tandem reaction forms three C-C bonds: one during the acylation and two during the Diels-Alder reaction. The products we obtained in our tandem reaction can be explained by the sequence intermolecular acylation between the N,O-keteneacetal and an acid chloride forming a C-C bond first followed by an intramolecular Diels-Alder reaction or by the alternative sequence: intermolecular Diels-Alder reaction first followed by intramolecular acylation second (figure 2).
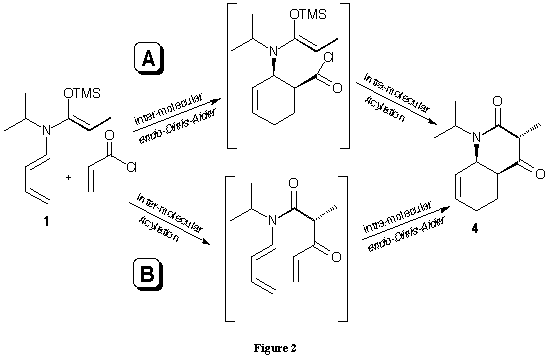
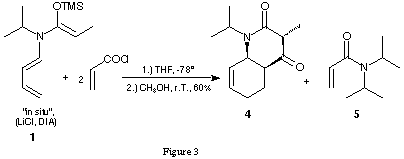
We prefer this second sequence because of the high diastereoselectivity of our tandem reaction: only one diastereoisomer was formed. In order to study the scope and limitations of this new tandem process it was important to synthesise a series of stable and isolable derivatives of N-butadienyl-N-isopropylketene-N,O-silylacetal of propionamide. We had used the trimethylsilylacetals created in situ for the application in our tandem reaction. Serious disadvantages of this procedure are the sensitivity of the ketene trimethylsilylacetal toward hydrolysis and the formation of the diisopropylamide 5 by the reaction of the acid chloride with the diisopropylamine formed from LDA (figure 3).
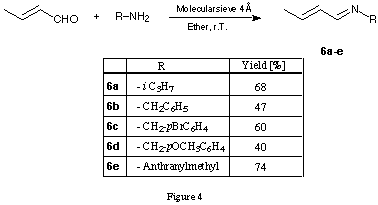
A series of N-alkyl-N-butadienyl amides were synthesised using the two step procedure: imine formation 6a - e catalysed by molecular sieves18 (figure 4) followed by base catalysed acylation with the corresponding acid chloride 19(figure 5).
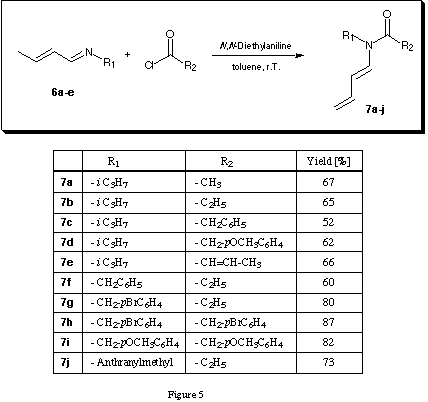
The N-alkyl-N-butadienyl amides 7a - j were obtained in good to excellent yields starting from crotonaldehyde, the alkyl amine and the corresponding acid chloride. We decided to try to synthesise the N-butadienyl-N-isopropylketene-N,O-tert.-butyldimethylsilylcetal of propionamide first which should be considerably more stable than the trimethylsilyl derivatives. The in situ deprotonation silylation which had been highly successful for the synthesis of the N-butadienyl-N-isopropylketene-N,O-trimethylsilylacetal of propionamide 112,13 could not be applied for the synthesis of the ketene-N,O-tert.-butyldimethylsilylacetal. Only after considerable optimisation of the reaction conditions following the prescription described by Rathke 20 good and reproducible yields of the N-butadienyl-N-isopropylketene-N,O-tert.-butyldimethylsilylcetal of propionamide 8a - j could be obtained (figure 6).
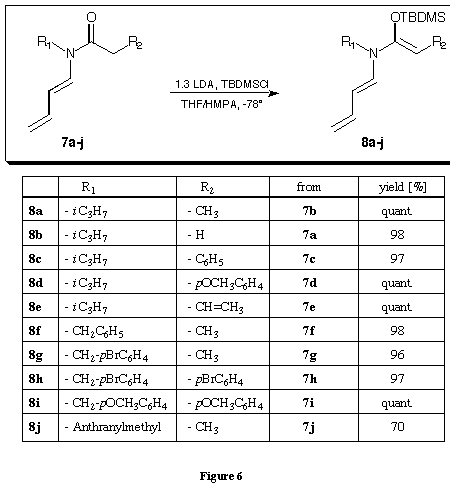
The amide 7a - j was deprotonated in the solvent mixture THF/HMPA = 10 : 1 at - 78deg.C with LDA. After 10 minutes the tert.-butyldimethylsilyl chloride dissolved in THF was added at the same temperature. The reaction mixture was allowed to warm to room temperature, diluted with pentane. Extraction against brine and evaporation of the solvent in the rotavap allowed to isolate the N-butadienyl-N-isopropylketene-N,O-tert.-butyldimethylsilylcetal of propionamide 8a - j in almost quantitative yield and in high purity as judged from the spectra. The synthesis of the N-para-bromobenzyl 8h and N-anthranylmethyl derivative 8j were not very satisfactory. Even though we usually used a 30 % excess of LDA for the deprotonation, the N-para-bromobenzyl derivative 8h was only silylated to the extend of 70 % whereas 30 % were still present as starting material. Submitting this mixture a second time to the deprotonation silylation conditions allowed to transform the starting material completely. The formation of different side products yielded a ketene-N,O-tert.-butyldimethylsilylacetal 8h of unsatisfactory purity. In the case of the N-anthranylmethyl derivative only a 70 % of the ketene-N,O-tert.-butyldimethylsilylacetal 8j could be isolated. Increasing the amount of LDA did not lead to an increased yield of the ketene-N,O-tert.-butyldimethylsilylacetal 8k. The relatively low yield could be due either to the competitive deprotonation in the benzylic position or to an electron transfer from the enolate anion to the aromatic system. Deuteration of the enolate with mono-deuteromethanol allowed to isolate the mono-deuterated starting material in 67 % yield and with a deuteration degree of 86 %. Within the precision of the NMR determination no deuteration at the benzylic position could be observed. This observation taken together with the fact, that treating of the N-anthranylmethyl derivative with LDA was the only deprotonation leading immediately to a deep greenish red coloured solution were taken as arguments favouring the electron transfer as side reaction diminishing the yield of the ketene-N,O-tert.-butyldimethylsilylacetal 8k. With the exception of the ketene-N,O-tert.-butyldimethylsilylacetals 8h and 8j the other tert.-butyldimethylsilylacetals 8a - g and 8i were obtained in excellent yield and high purity, free of diisopropylamine. The tert.-butyldimethylsilylacetals 8a - j could be purified via extraction and could be stored over months in the refrigerator. However trials to purify the ketene-N,O-acetals 8a - j via chromatography lead to hydrolysis. Even the use of deactivated silicagel or aluminium oxide allowed only the isolation of the hydrolysed starting material.
Acylation reactions of ketene-N,O-acetals are usually catalysed by Lewis acids. In our tandem process no Lewis acid had been added. The LiCl present in the reaction mixture using the ketene-N,O-trimethylsilylacetal 1 synthesised in situ could possibly act as a weak Lewis acid. Using the pure ketene-N,O-tert.-butyldimethylsilylacetal 8a free of LiCl and diisopropylamine and submitting it to the reaction conditions optimised for the ketene-N,O-trimethylsilylacetal the bicyclic product 4 could be obtained in 50 % yield (figure 7).
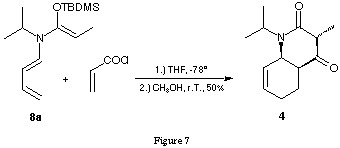
Work-up and crystallisation were considerably facilitated by the fact that no acroyldiisopropylamide 5 had been formed as side product. This result clearly indicates that the presence of LiCl is not necessary for the success of the tandem reaction.
Crotonyl chloride or methacryl chloride could also be used as dienophiles for the tandem reaction (figure 8).
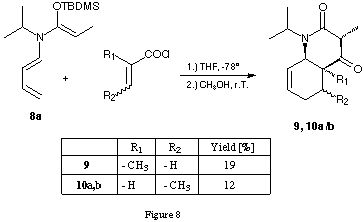
The TLC-analysis of the reaction mixture indicated that during the reaction several side products were formed. The chromatographic isolation of the bicyclic products 9, 10a/b proved to be tedious and time consuming. Using methacryl chloride as dienophile one single diastereoisomer 9 was obtained. In the case of the crotonyl chloride, the starting material was a 4 : 1 mixture of the (E)- and (Z)-diastereoisomer. The product obtained after the reaction of crotonyl chloride with the ketene-N,O-tert.-butyldimethylsilylacetal 8a was a mixture of the diastereoisomers 10a and 10b at the position C(5). One of the two diastereoisomers 10a could be crystallised directly from the mixture. The relative configuration of the tandem products was determined with the help of NOESY-spectra. The cis-junction of the two rings was evident from the cross-peaks between the H at the bridgehead positions. The bicycles are in a half-chair/boat conformation locked by the isopropyl group of the lactame nitrogen (figure 9).
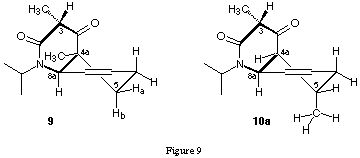
The cross-peaks between the methine H at C(3) and the olefinic H at C(8) as well as the cross-peaks between the H at C(8a) and the pseudo-axial H of the methylene group at C(5) are only compatible with such a locked conformation. A further indication for this strong conformational preference came from the deuterium exchange experiment (figure 10).
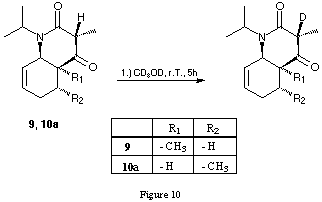
Dissolving the bicyclic products in deuteromethanol and observing the 1H-NMR spectrum during 5 h at room temperature showed complete exchange of the H at C(3), keeping the configuration at C(3) intact. The isolated product must therefore be the thermodynamically preferred diastereoisomer.
In conclusion, a series of N-butadienyl-N-alkylamides 7a - j have been synthesised and a method has been developed to obtain the stable, storable ketene-N,O-tert.-butyldimethylsilylacetals 8a - j in excellent yield. This ketene-N,O-acetals undergo the tandem reaction Diels-Alder reaction followed by acylation in good to moderate yield using acryloyl, crotonyl or methacryl chloride. The bicyclic products 4, 9, 10a/b are formed in high chemo- and diastereoselectivity by this tandem process.
We acknowledge financial support from Swiss National Science Foundation.
Experimental Part:
General . All reactions were carried out under Ar. Solvents were dried by distillation from drying agents as follows: THF (Na), Et2O (CaH2), CH2Cl2 (CaH2), MeOH (Mg), EtOH (Mg). Silica Gel 60 (Merck) was used for flash-chromatography (FC). M p were determined in open capillary tubes on a Kofler melting point apparatus (Thermovar, C. Reichert AG, Vienna) and are uncorrected. IR spectra: Perkin Elmer 1720 X FT IR spectrophotometer using liquid films between potassium chloride discs or in CHCl3,or CCl4 unless otherwise noted. NMR spectra: 1H at 400 MHz on a Bruker AMX 400, at 200 MHz on a VARIAN Gemini 200.13C 100 MHz on a Bruker AMX 400, 50 MHz on a VARIAN Gemini 200. If not otherwise mentioned spectra were measured in CDCl3 with CHCl3 as internal standard. MS: The HRMS were measured on aVacuum Generator Micromass 7070E and the normal MS on a NERMAG R30-10 (70 eV); relative peak intensities are given in % of the base peak (= 100%). Microanalyses were performed in the microanalytical laboratories of CIBA-GEIGY Ltd., Marly/Fribourg.
Typical Experimental Procedures and selected Data's:
N-Isopropyl-(E)-2-butene-1-imine (6a):
To a cold (0deg.C) solution of crotonaldehyde (14.02 g, 0.2 mol) and isopropylamine (11.82 g, 0.2 mol) in Et2O (70 ml) was added in small portions molecular sieve 4Å (80 g). After addition was complete the mixture was stirred during 4h at r.t.. The mixture was filtered and the molecular sieve was washed with Et2O (4 x 60 ml). Concentration of the filtrate and distillation (29 deg.C/11 Torr) of the crude product afforded the imine 6a (15.3 g, 68 %) as a colorless oil.
IR (film): ν = 2970, 2940, 2830, 1660, 1625s, 1465, 1450, 1380, 1320, 1160, 980, 960, 930 cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.85 (d, 1H, J = 7.7 Hz), 6.22-6.13 (m, 2H), 3.30 (sept,1H, J = 6.2 Hz), 1.91 and 1.88 (d, 3H, J = 5.2 Hz), 1.17 (d, 6H, J = 6.3 Hz).
13C NMR (50 MHz, CDCl3): δ=159.9, 139.7, 132.2, 60.9, 24.1, 18.2.
MS (EI): m/z = 112 (16, M + 1+), 96 (100), 94 (10), 79 (14), 68 (27), 55 (25).
(E)-N-Isopropyl-N-propionyl-1-amino-1,3-butadiene (7b):
To a solution of propionylchloride (10.18 g, 0.11 mol) and N,N-diethylaniline (17.9 g, 0.12 mol) in dry toluene (30 ml) was added a solution of imine 6a (12.0 g, 0.11 mol) in toluene (30 ml) dropwise at r.t. under Ar. After addition was complete the mixture was stirred over night at this temperature. The mixture was filtered over Celite and the residue was washed with toluene (2 x 50 ml). The combined organic layers were combined and washed with 1M aq. HCl solution (2 x 100 ml), sat. aq. NaHCO3 solution (2 x 100 ml) and H2O (2 x 100 ml). The organic phase was dried (MgSO4) and evaporated under reduced pressure. The residue was purified by distillation (51 deg.C/0.03 Torr) to give 7b (10.86 g, 65 %) as a light yellow oil.
IR (CCl4): ν = 2980, 2940, 2880, 1680, 1635, 1605, 1460, 1430, 1400, 1380, 1365, 1340, 1290, 1250, 1220, 1170, 1130, 1075, 1000, 925, 890 cm-1.
1H NMR (400 MHz, CDCl3): δ = 6.43 (d, 1H, J = 13.7 Hz), 6.33 (dxt, 1H, J = 16.9, 10.4, 10.1 Hz), 6.00 (br s, 1H), 5.20 (d, 1H, J = 16.9 Hz), 5.08 (d, 1H, J = 10.1 Hz), 4.66 (br s, 1H), 2.39 (q, 2H, J = 7.3 Hz), 1.24 (d, 6H, J = 6.9 Hz), 1.12 (t, 3H, J =7.3 Hz).
13C-NMR(100 MHz, CDCl3): δ = 172.4, 134.4, 128.8, 123.0, 115.8, 46.5, 28.0, 19.8, 9.2.
MS (EI): m/z = 168 (22, M+ + 1), 167 (M+), 153 (6), 112 (25), 111 (33), 98 (14), 97 (75), 96 (100), 82 (13), 70 (27), 69 (46), 68 (33), 58 (23), 57 (45), 44 (40), 43 (52).
(Z)-N-[(E)-Buta-1,3-dienyl]-N-isopropyl1-[(tert-butyl-dimethylsilyl)-oxy]prop-1-enamine (8a):
In a flamedried three-necked flask fitted with magnetic stirrer, septum, argon bubbler and thermometer, a 1.6M solution of n-BuLi (hexane, 3.0 ml, 4.2 mmol) was added dropwise to a solution of anh. (i-Pr)2NH (0.6 ml, 4.3 mmol) in dry THF (10ml) at -78 deg.C. After addition was complete the mixture was allowed to reach 0 deg.C and stirred 30 min at this temperature. The mixture was cooled to -78 deg.C and freshly distilled HMPA (1.0 ml) was added in one stream to the solution. Afterwards a solution the dienamide 7b ( 0.47 g, 3.0 mmol) in dry THF (2 ml) was added slowly at the same temperature and stirring was continued for 10 min at -78 deg.C. To this solution was added dropwise a solution of TBDMSCl (0.51 g, 3.4 mmol) in dry THF (2ml) at -78 deg.C. After addition was complete, the mixture was warmed up to r.t. and stirred for 2h.After stirring pentane was added (30 ml) and the resulting solution was washed with water (2 x 10 ml) and with saturated. aq. NaCl solution. (2 x 10 ml). The org. phase was dried (Na2SO4) and the solvent was removed to give pure silylenolether 8a (0.86 g, quant.).
IR (CCl4): 3085, 3045, 2960, 2930, 2860, 1670, 1630, 1470, 1460, 1320, 1255, 1050, 995 cm-1.
1H NMR (200 MHz, CDCl3): δ = 6.23 (dxt, 1H, J =16.5, 10.6, 10.5), 6.18 (d, 1H, J = 14.1), 5.32 (dd, 1H, J = 13.7, 10.6 Hz), 4.75 (dd, 1H, J = 16.8, 2.2 Hz), 4.53 (dd, 1H,J = 10.3, 2.1 Hz), 4.33 (q, 1H, J = 6.7 Hz), 3.59 (sept, 1H, J = 6.7 Hz), 1.56 (d, 3H, J = 6.7 Hz), 1.22 (d, 6H, J = 6.7 Hz), 0.93 (s, 9H), 0.1 (s, 6H).
13C NMR (100 MHz,CDCl3): δ = 147.9, 138.1, 138.0, 106.6, 104.1, 99.6, 50.5, 26.4, 20.9, 18.8, 11.7, -3.5.
MS (EI): m/z = 282 (13, M+ + 1), 281 (19, M+), 267 (16), 266 (22), 115 (8)), 75 (13), 74 (17), 73 (100).
(3RS,4aSR,8aRS)-1,2,3,4,4a,5,6,8a-Octahydro-1-sopropyl-3-methyl-chinoline-2,4-dione (4) 12
In a flamedried three-necked flask fitted with magnetic stirrer, septum, argon bubbler and thermometer a solution of silylenolether 8a (0.8 g, 2.9 mmol) in dry THF (15 ml) was treated dropwise with acryloylchloride (0.3 ml, 3.5 mmol) at -78 deg.C. After addition was complete the mixture was stirred for 2h at this temperature and allowed over night to reach r.t. The mixture was hydrolized with MeOH (5 ml) and the solvent was removed by evaporation under reduced pressure. The residue was dissolved in CHCl3 (40 ml) and washed with 1M aq. HCL soln (2 x 20 ml) and with saturated aq. NaCl solution (2 x 20 ml). The organic layer was dried (Na2SO4) and the solvent was removed to liberate the crude material, which was purified by flash column chromatography on silica gel (EtOAC/hexane, 1:1) to furnish the product 4 (0.32 g, 50 %) as a yellow oil which could be crystallized. (m p 79 deg.C; Lit. 80 deg.C 12.
References
1 E. Leete, Planta. Med. 1990, 56, 339.
2 E. E. van Tamelen, J. Willet, R. B. Clayton, K. E. Lord, J. Am. Chem. Soc. 1966, 88, 4752.
3 R. Robinson, J. Chem. Soc. 1917, 762.
4 R. Robinson, J. Chem. Soc. 1917, 876.
5 R. B. Ruggeri, K. F. McClure, C. H. Heathcock, J. Am. Chem. Soc. 1989, 111, 1530.
6 S. D. Knight, L. E. Overman, G. Pairaudeau, J. Am. Chem. Soc. 1993, 115, 9293.
7 L.F. Tietze and U. Beifuss, Angew.Chem., 1993, 105, 137;
Angew.Chem Int.Ed. Engl.
1993, 32, 131.
8 T.L. Ho, Tandem Organic Reactions, Wiley, New York, 1992.
9 S. Huber, P. Stamouli and R. Neier, J.Chem.Soc., Chem.Comm., 1985, 533.
10 J. Schoepfer, E. Eichenberger and R. Neier, J.Chem.Soc., Chem.Comm., 1993, 246.
11 S. Huber, P. Stamouli, T. Jenny and R. Neier, Helv.Chim.Acta, 1986, 69, 1898.
12 M. Baak, Y. Rubin, A. Franz, H. Stoeckli-Evans, L. Bigler, R. Nachbaur, R. Neier, Chimia 1993, 47, 233.
13 M. Baak, 'Doktorarbeit', Institut für Organische Chemie, Universität Fribourg, 1991.
14 T. Gallagher, P. Magnus, J. C. Huffman, J. Am. Chem. Soc. 1982, 104, 1140.
15 P. Magnus, P. Pappalordo, I. Southwell, Tetrahedron 1986, 42, 3215.
16 P. Magnus, N. L. Sear, Tetrahedron 1984, 40, 2795.
17 S. F. Martin, W. Li, J. Org. Chem. 1989, 54, 268.
18 K. Taguchi, F. H. Westheimer, J. Org. Chem. 1971, 11, 1570.
19 W. Oppolzer, L. Bieber, E. Francotte, Tetrahedron Lett. 1979, 11, 981.
20 R. Woodbury, M. Rathke, J. Org. Chem. 1977, 43, 881.