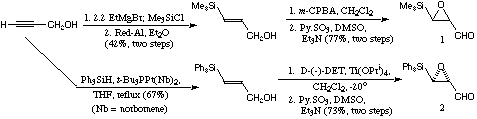
Figure 1
The alpha-beta-epoxyaldehydes 1 and 2 were prepared simply, as shown in Fig. 1, 1 was prepared only in the racemic form, and 2 was prepared in enantiomerically enriched form via Sharpless enantioselective epoxidation. It is interesting to compare the preparations of the two intermediate vinylsilane alcohols, the catalytic hydrosilylation is far more convenient and higher yielding than the standard method used for the trimethylsilyl compound (the work with this compound was carried out before we completed the work on Pt(0) catalysed hydrosilylation of propargyl alcohols).[9]
Figure 1
The results of addition reactions of both 1 and 2 are presented in Fig. 2 and Table 1, and are unexpected in view of the previous results with alpha-beta-epoxyaldehydes which lack a substituent cis- to the aldehyde (vide supra). Similar findings have been reported for epoxyaldehyde 1 by Sato et al.[10] Our results, which are reported in the table in Fig. 2, suggest that steric demands in both the aldehyde and the nucleophile are important in determining the stereochemical outcome of additions to 1 and 2; in general additions to the trimethylsilylepoxide 1 are somewhat less stereoselective than those to the triphenylsilylepoxide 3.
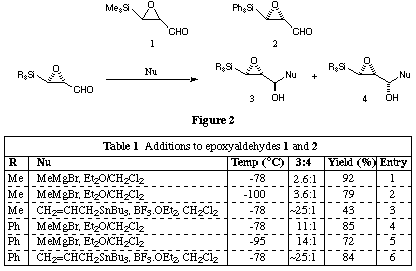
The observation of any useful level of diastereoselectivity in the addition of simple nucleophiles to trans-substituted alpha-beta-epoxyaldehydes was unexpected, given the lack of diastereoselectivity found when a C-substituent replaces the silyl group. Nevertheless, the sense of the diastereoselectivity observed in additions to 1 and 2 is consistent with a `non-chelation' model, similar to the working hypothesis in our group used to account for the results from cis-alpha-beta-epoxyaldehydes.[11] The increase in diastereoselectivity on changing from a trimethylsilyl to a triphenylsilyl group (Table 1, entries 1-3 vs. 4-6), and the relatively high diastereoselectivity observed in the addition of allyltri-n-butylstannane (Table 1, entries 3 and 6) also deserve comment.
It is not easy to find an electronic explanation for the diastereoselectivity increase on changing from a carbon- to a silyl-substituent. We suggest that one rationalisation which might not be unreasonable is to propose that a steric effect of the silicon substituent, and a preferred conformation of the alpha-beta-epoxyaldehyde, combine to provide the directing effect. Our working hypothesis concerning the `non-chelation controlled' addition of nucleophiles to alpha-beta-epoxyaldehydes and imines[12] is based on the work of others on the preferred conformations of simple alpha-beta-epoxyaldehydes,[13] and on the interpretation of our experimental results (and those of others). In our model we assume that of the two conformations which allow pi orbitals of the C=O group to mix with the Walsh orbitals of the three-membered ring, A and B (Fig. 3),[14] dipole considerations would lead to A being favoured. In the transition state where a build-up of charge on the oxygen atom of the reaction C=O group is expected, this dipole-dipole repulsion will presumably be increased.
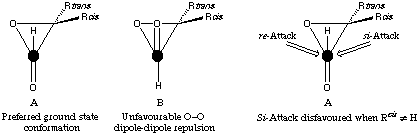
Figure 3
If Rcis is not a hydrogen, then attack on the si-face of the aldehyde will be disfavoured because of the steric hindrance of Rcis. Presumably, this effect would be greater for an addition with a late transition state, and this might be the origin of the relatively high diastereoselectivity observed for addition of the uncharged allylstannane, compared with the Grignard reagent.
Simple molecular mechanics calculations using MacroModel and the two model epoxyaldehydes shown in Figure 4 predict the minimum energy conformation of these systems to be in the region Ø~ 100-110 degrees (Ø defined in Fig. 4, and energy calculated at 10 degree intervals). Taking the most simplistic structural parameters, this dihedral angle would be expected to be ~140-150 degrees in our working hypothesis. While these theoretical calculations do not place the aldehyde in exactly the same orientation as in our working model, they do suggest that the energy increases rapidly as Ø approaches zero, where the alpha-C-O bond of the epoxide and the C=O bond are aligned parallel.
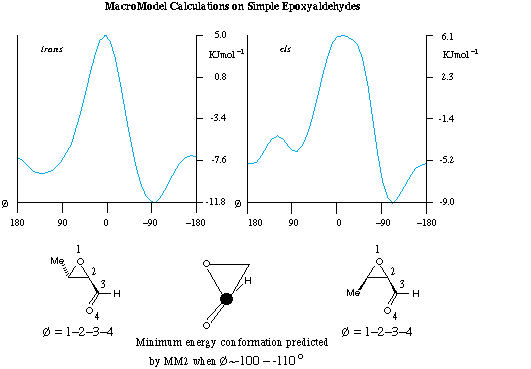
Figure 4
In view of the preceding results, we also examined alpha-beta-epoxyaldehydes whose X-ray crystal structures have been determined. A search of the Cambridge Crystallographic Database identified two such structures, representations of which are provided in Figure 5.[15] Both of these have dihedral angles, as defined above, which are not inconsistent with the foregoing discussion. However, both of these are far from ideal, being heavily substituted, and in the case, an oxygen atom (OA) of a carbonyl group appears to interact with the aldehyde carbonyl group.
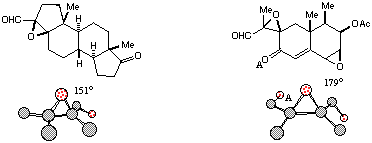
Figure 5
The triphenylsilyl-substituted alpha-beta-epoxyaldehyde 2 is crystalline, and we were successful in growing a crystal suitable for single crystal X-ray diffraction studies.[16] The asymmetric unit of this crystal contained two independent molecules (labelled A and B), which differed by small changes of the triphenylsilyl group. Pictorial representations of the results of this crystal structure analysis are presented in Figure 6. The dihedral angle Ø is essentially the same in each independent molecule, averaging out at 167 degrees.
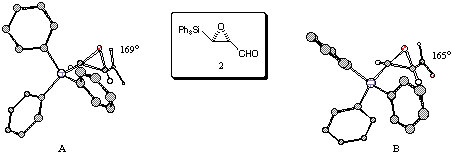
Figure 6
It appears that the preceding modelling and results from measurements in the solid state are not inconsistent with the suggestion outlined in Figure 3 that dipole-dipole repulsion is a significant factor in determining the minimum energy conformation of alpha-beta-epoxyaldehydes of this type. Given this, and the assumption that similar effects might be operating in the transition state for nucleophilic attack under non-chelating conditions, it is possible that the very large size of the triphenylsilyl group might be responsible for (as yet undefined!) steric interactions which lead to the diastereoselectivity observed in additions to these systems.