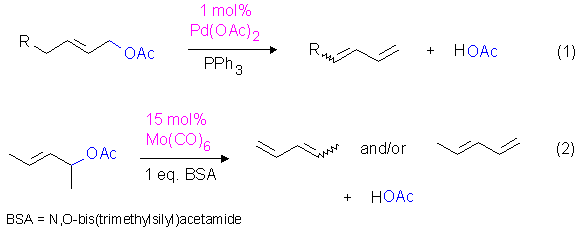
Some transition metal catalysts such as palladium and molybdenum complexes have already been shown to be effective in the preparation of 1,3-dienes from allylic acetates (Equations 1 and 2).[2]
At least in the palladium cases, pi-allyl intermediates [click here for mechanistic details] are obviously involved as proposed by Tsuji et al..[2a] Even saturated tertiary alcohols and esters could selectively be converted into alkenes using molybdenum catalysts.[3] Although these reactions are comparable with respect to the products formed, they must be mechanistically different as allylic intermediates cannot be involved in the latter cases.
For example, the tricyclic isomeric sesquiterpenes alfa- and beta-cedrene could be prepared from the corresponding tertiary acetate. Kinetic rather than thermodynamic factors obviously govern the selectivity (vide infra), so that predominantly the thermodynamically less stable exo methylene compound beta-cedrene could be obtained. Gas phase pyrolysis of esters or amine oxides [4] are the only general preparative alternatives leading to a similar product distribution but they are technically more complicated than our procedure.
In connection with our research on transition metal complexes of unsaturated carbonyl compounds,[5] we became interested in convenient new syntheses for potential 1-oxa-1,3-diene ligands. In this paper, a simple and selective two-step preparation of alfa,beta-unsaturated ketones from inexpensive starting materials based on the Aldol condensation is reported.
The Aldol condensation,[6] although being a simple and powerful tool for the preparation of unsaturated carbonyl compounds, may suffer from poor regioselectivity in the dehydration step. As an example, aldols with alkyl substituents in the gamma-position usually lead to mixtures of alfa,beta- and beta,gamma-unsaturated ketones under thermodynamic control. Thus, the final product purification normally requires careful and time consuming distillation procedures to separate the product isomers.
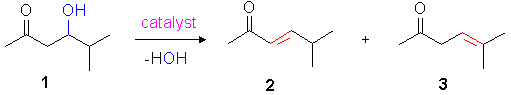
In our synthetic approach, the beta-hydoxyketones are formed in the first step by conventional Aldol addition. The beta-hydroxyketone 1, for example, is easily obtained from iso-butyric aldehyde and acetone in 80% yield. When dehydrated thermally without a catalyst, only an 18% conversion was achieved even at 120°C, the unsaturated ketones 2 and 3 were formed in a 3 : 1 ratio (entry 1, Table 1a). At 80°C, where all the metal catalysed reactions were performed, the thermal dehydration is hardly proceeding at all (4% conversion, entry 2) and can thus be neglected as competing reaction. Acid catalysis, catalytic amounts of iodine and even "neutral" alumina do catalyse the dehydration effectively at that temperature (entries 3-6), but the regioselectivity is unacceptably low. In the iodine case (entry 5 in pink), a 1 : 1 mixture of 2 and 3 is obtained (complete thermodynamic control), thus indicating almost identical thermodynamic stabilities of these products. This latter experiment has, diffently from all others, been performed in an NMR-tube in order to avoid corrosion of the autoclave steel. The product ratio has been determined by proton NMR spectroscopy in this case.

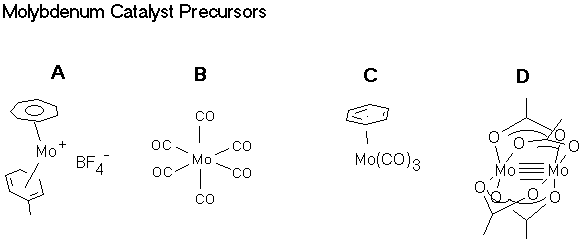
Results completely different from the above-mentioned are obtained when molybdenum compounds such as the cycloheptatrienyl complex A, hexacarbonyl molybdenum (B), benzene tricarbonylmolybdenum (C) or molybdenum acetate (D) are employed as catalyst precursors (click here if you want to take a look at the structures of these complexes). In all these cases (entries 7-10, Table 1b) a markedly increased regioselectivity for the formation of the conjugated 1-oxa-1,3-diene 2 is observed. Total conversion of the starting material could, however, not be achieved in these runs (blue numbers in Table 1b, ranging from 19 to 62 %). Furthermore, when molybdenum carbonyl is used as the metal source, the addition of a relatively large excess of acetic acid, based on the metal compound (at least 10 : 1, usually 20 : 1), is necessary for the reaction to proceed at an acceptable rate.

In order to improve on conversion, the addition of magnesium sulphate monohydrate turned out to be essential (entries 11 and 12). At best (entry 11 in red), with only 0.5 mol% of complex A , the 1-oxa-1,3-diene 2 is obtained almost exclusively at 97% conversion. This reaction could be repeated on a 100 mmol scale with identical yield and product distribution. Finally, it should be noted that diethyl ether was used as solvent merely to guarantee a simple workup procedure. The dehydrations also proceed analogously in dioxane under identical conditions without the use of an autoclave. The isolation of the oxadiene then, however, requires subsequent removal of the solvent by careful distillation.
As obvious from the iodine catalysed dehydration reaction (entry 5, Table 1a) proceeding under thermodynamic control, the conjugated and non-conjugated enones 2 and 3 are of very similar stability. In order to select further applications of our reaction sequence, we intended to predict the stability of the dehydration products on the basis of calculations. This would enable us to select reactions where a severe competition in the formation of product isomers could be expected under thermodynamic control, thus allowing us to test for improvement in a metal catalysed reaction.
Surprisingly, several types of semiempirical calculations [7] summarised in the upper half of Table 2 all predict the beta,gamma-unsaturated ketone to be more stable by up to 18 kJ/mole, which is not in agreement with the experiments. Geometrical as well as energetical results from calculations performed by using the C.I. option of MOPAC do not significantly differ from those obtained without such CI treatment . For calculational details, click here. With the AM1 and PM3 method, both oxadienes are predicted to be essentially planar regarding the ketone and alkene pi fragments. Only the MNDO calculations lead to non-coplanar arrangements for the pi fragments of both isomers. This difference has, however, no severe consequences for the calculated enthalpies of formation, which are very similar in MNDO and AM1 calculations. If you are interested in these non-planar geometries at the MNDO level, take a look at the molecules 2(MNDO) and 3(MNDO).
Even "low-level" ab initio calculations,[8] e.g. using the 3-21G basis set correctly predict 2 and 3 to be quite similar in ground state energy. There is a slight preference for the conjugated 1-oxadiene that further decreases when larger basis sets are employed an electron correlation is introduced.
Two additional coplanar conformers of 2 and 3, i.e. 2' and 3', being rotamers with respect to the alfa-carbonyl bond, have explicitly been investigated in semiempirical as well as in RHF and MP2 calculations employing the 3-21G basis set. They were found to be significantly higher in energy than 2 and 3, respectively. If interested in these data, click here.
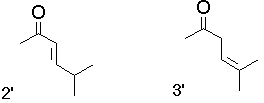
The relative stability of the regioisomers 2 and 3 is found to be almost identical if either the 3-21G or the 6-31G* basis set is employed as long as electron correlation is included in these calculations using the MP2 method. For further details on the ab initio calculations, click here.
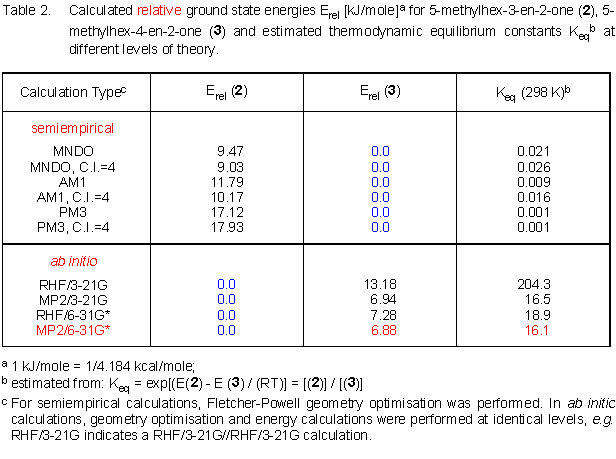
The preference for the conjugated isomer from MP2/6-31G* calculations amounts to about 6.9 kJ/mole (bottom part of Table 2). As a raw estimate for the thermodynamic product ratio, equilibrium constants calculated for 298 K are also given in Table 2. The value of about 16 from the MP2/6-31G* results is still relatively large compared to the experiments. The absolute energies obtained in the semiempirical and ab initio calculations are given in Table 3a and 3b, respectively. Furthermore, the ab initio geometries (pdb data format) of 5-methylhex-3-en-2-one (2) and 5-methylhex-4-en-2-one (3) at the MP2/6-31G*//MP2/6-31G* level are available. Most remarkably about these geometries, even for the beta,gamma-unsaturated ketone an almost coplanar arrangement of ketone and alkene moieties is found.
Neither thermal nor acid or iodine catalysed dehydration of 4-hydroxy-5-methylhexan-2-one (1) provides access to the pure conjugated 1-oxa-1,3-diene 2. Under thermodynamic control, a 1:1 mixture of conjugated and non-conjugated isomer is obtained. Thus the stabilisation arising from oxadiene conjugation (in 2) and alkene trisubstitution (in 3) ballance each other. Using semiempirical methods, stabilisation of the alkene by alkyl substitution is overestimated with respect to the delocalisation in the oxadiene pi system, leading to a preference for 3 regarding the heats of formation. Contrary to this, ab initio calculations predict 2 to be more stable. Whereas the energy difference is relatively large at the RHF/3-21G level (13 kJ/mole), it drops with larger basis sets and the introduction of electron correlation to 6.9 kJ/mole at the MP2/6-31G* level. After additional correction for the difference in zero point vibrational energy (ZPE), the energy differences of 2 and 3 on both MP2 levels decreases to the same value of about 3.7 kJ/mole, leading to an estimated equilibrium constant of 4.5. This is in very good agreement with the experimental product ratios between 1 and 3 for the acid and iodine catalysed reactions. A prediction of relative product stabilities which quantitatively represents the experimental findings thus requires quite sophisticated and expensive calculations. Semiempirical calculations are obviously not sufficient to obtain even qualitative results on the thermodynamic stability of these products. It has, however, to be seen from a larger number of calculations on differently substituted oxadienes, if semiempirical calculations probably favour the non-conjugated isomer by a constant energy so that an empirical correction of these energies would allow to obtain at least qualitatively correct results for these relative stabilities.
In the metal catalysed reactions, a definite preference for the formation of the conjugated isomer 2 is found. The low selectivity observed with molybdenum acetate is possibly due to the low solubility of this compound in ether leading to some kind of heterogeneous reaction. When molybdenum acetates are formed in situ from hexacarbonyl molybdenum and acetic acid, presumably different, soluble species are generated. The role of the magnesium sulphate in entries 11 and 12 of Table 1b is not totally understood, it could either function as drying agent and remove the water formed during the dehydration. Alternatively, it could activate the catalyst by acting as support. At the moment, due to the relatively large amounts of magnesium sulphate needed for optimal conversion (40 - 110 mol%), its function to bind the water seems more likely. The conversion of precursor A into an active catalyst is obviously much more efficient than the analogous reaction of hexacarbonyl molybdenum. Therefore, only 0.5 mol% of the cationic cycloheptatrienyl complex are sufficient to completely convert the starting material in contrast to 2 mol% of the hexacarbonyl.
As a mechanistic explanation for the observed dehydration regioselectivity, formation of hydroxo-metal intermediates and subsequent abstraction of a proton from the sterically less hindered, i.e. alfa-ketone position, can be proposed. In a primary step, insertion of the molybdenum into the C-O bond can be envisioned. Scheme 2 shows a possible catalytic cycle for such a reaction, finally regenerating the active catalyst by reductive elimination of water.
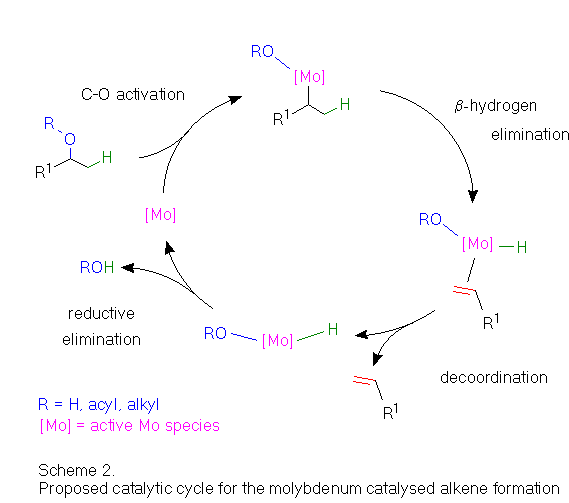
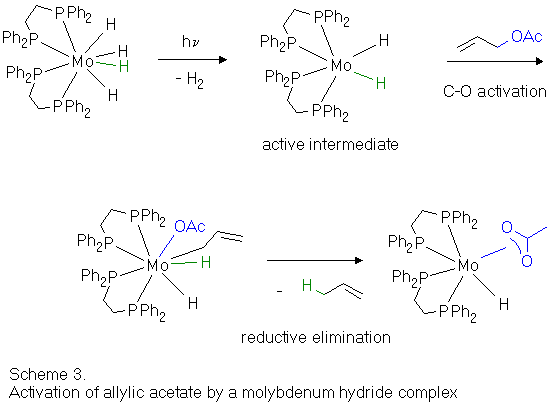
A similar reaction (Are you interested in details ? (Scheme 3)) involving C-O activation of allylic acetate has already been observed for a photochemically generated, coordinatively unsaturated molybdenum hydride complex.[9] However, reductive elimination involving the allyl ligand subsequently occurs in that case instead of a beta-hydrogen abstraction as proposed in the above catalytic cycle (Scheme 2).
Due to steric bulk at the metal center, a preference for proton transfer from the less hindered position of the substrate to one hydroxo or carboxylato ligand can be expected to be more facile, leading to the less substituted alkene. This kind of selectivity has already been observed, e.g. in the formation of beta-cedrene ( vide supra) from tertiary alcohols and esters.
A cyclic transition state as shown in Equation 5 could also be envisioned, assuming a simultaneous transfer of the hydroxy group and the hydrogen in the beta position. Especially when acetates are involved in the elimination reaction, e.g. in the cedrene case, a very crowded transition state would result. This might help in the selection of the least hindered beta position for hydrogen transfer. Additional support for the above mechanistic hypothesis comes from the recent observation [10] that a scrambling of carboxylate ligands on dimolybdenum centers, e.g. between different dimolybdenum tetracarboxylates, is a very facile process even at room temperature. Such a reaction would be an important part of a process as indicated below, where simultaneous transfer of a "blue" OR fragment to and a "black" one from the metal center occurs.
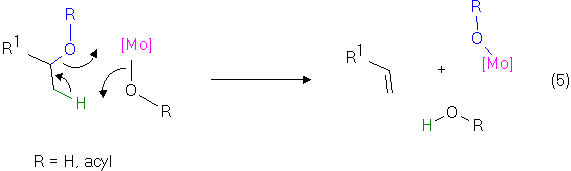
Besides from the example discussed here, we already applied the above preparative sequence to several other dehydration reactions to form alfa,beta-unsaturated ketones predominantly (e.g. 1-cyclopropyl-4-methylpent-2-en-1-one) or exclusively (4-methylpent-3-en-2-one). We are still investigating scope and limitations of the reaction in more detail.
The author thanks the Max-Planck-Gesellschaft and the Fonds der Chemischen Industrie e.V. for financial support. Calculations have been performed at the Regionales Rechenzentrum der Universität zu Köln. I am especially grateful to this organisation for a generous allotment of computer time and helpful cooperation. Indispensable technical support from Karlheinz Boll at the computing center of the Max-Planck-Institut für Kohlenforschung in setting up the WWW environment is gratefully acknowledged.
Hexacarbonyl molybdenum was obtained from FLUKA and used as received, all other catalyst precursors were prepared according to ref. [3]. 4-Hydroxy-5-methylhexan-2-one was prepared by a modification of a published procedure [11] from iso-butyric aldehyde and acetone in the presence of 2.5 N aqueous sodium hydroxide and obtained in 80% yield after distillation. [12]
The beta-hydroxyketone (10 mmol) was dissolved in 30 ml of diethyl ether. The solution was placed in the glass insert of a 100 ml stainless steel autoclave, catalyst and additives (see Table 1b for details) were added. The reaction mixture was then heated under argon to 80°C for 24 h. After cooling, the mixture was extracted once with saturated NaHCO3 solution to remove the molybdenum compounds from the organic layer. The latter was then washed with saturated NaCl solution, dried over magnesium sulphate, filtered and concentrated in vacuo. The crude products could be distilled in vacuo without isomerisation (G.L.C. and NMR control) as long as acid free glassware is used. Analytically pure unsaturated ketones were obtained. They were analysed by NMR and G.L.C. in order to determine the isomeric ratio. Click here for analytical data of compounds 1, 2 and 3.
[1] a) R.V. Hoffman, R.D. Bishop, P.M. Fitch and R. Hardenstein, J. Org. Chem., 1980, 45, 917-919; E. Keinan and Y. Mazur, J. Org. Chem., 1978, 43, 1020-1022.
[3] Th. Schmidt, Tetrahedron, 1991, 47, 8155-8160.
[4] S.P. Acharya and H.C. Brown, J. Org. Chem., 1970, 35, 196-206 and references therein.
[6] Review: A.T. Nielsen and W.J. Houlihan, Org. React., 1968, 16, 1-438.
[9] T. Ito, T. Matsubara, Y. Yamashita,J. Chem. Soc., Dalton Trans., 1990, 2407-2412.
[10] J.M. Casas, R.H. Clayton and M.H. Chisholm, Inorg. Chem., 1991, 30, 358-360.
[12] Th. Schmidt, Habilitation Thesis, University of Cologne, 1992.
8. Footnotes
Remark on Syntax
Up to now, I could not find a generally working possibility to
include non-standard characters, specifically greek letters, into my HTML-documents, so that different browsers will recognise them correctly. Therefore, I finally
decided on just emphasising the names of such characters by italics throughout this document, if not these
characters are part of an image (where this problem does not exist).
If anybody would like to comment on this or has some helpful advise, please
contact me by e-mail.
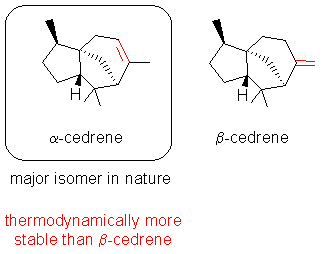
These tricyclic sesquiterpenes are the major ingredients of cedar wood oil, for example from the american cedar Juniperus virginiana L.. They were first isolated by Ph. Walter in 1841 (Liebigs Ann. Chem., 1841, 39, 247). Natural cedrene mainly consists of the alfa isomer, along with some of the tertiary alcohol cedrol, and a small amount of beta-cedrene. Artificial cedrene, as industrially produced and already obtained by Walter from cedrol and phosphoric acid is essentially pure alfa-cedrene.
All calculations were performed on IBM RS6000 and DEC Alpha /AXP 7000 systems of the University of Cologne Computing Center employing the MOPAC 93 [7] and the Gaussian 92 [8] programs.
Starting geometries for the semiempirical calculations were designed using standard bond lengths and angles. Arrangements with coplanar and orthogonal pi fragments were both explicitely tested. They were found to converge to the same stationary points in the course of the geometry optimisation. All optimisations were performed with the PRECISE keyword using the Fletcher-Powell method. In order introduce some extent of electron correlation correction, a 4x4 CI as resulting from the C.I.=4 option in MOPAC was employed.
Further details of the calculations are available from the author on request.

![[click here for mechanistic details]](sdt2.gif){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}