Since the discovery of a method to produce macroscopic amounts of fullerenes by Krätchmer and Huffman1 there has been an explosion of interest in this family of soluble closed-caged molecules composed entirely of carbon in its sp2-hybridized state. Many organic transformation reactions of C60 have been reported and distinct trends exist2. For example, C60 behaves as an electron-deficient olefin with fairly localized double bonds between the 6,6 junctions (in a [5]radialene-type bonding pattern), to which addition reactions may occur often in a reversible manner. Thus, oxidation occurs readily, but chemically the oxygen is not firmly bound and like many other derivatives, C60O shows a tendency to revert to C60 upon heating3.
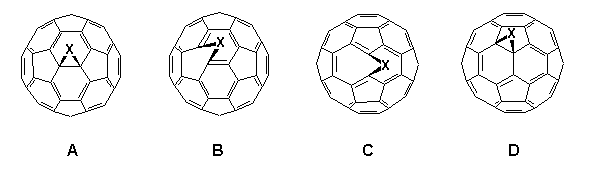
The preparation of stable, more synthetically useful mono-adducts of C60 is currently one of the prime objectives in the rapidly expanding area of fullerene chemistry. So far considerable success has been achieved in the building of three-3,4, four-5, five-6 and six-membered ring systems7 onto C60 and high-field 13C NMR spectroscopy is proving to be a powerful probe in elucidating their structure4,8,9. In theory, mono-adducts of C60 may possess one of four possible structures A-D depending upon whether attachment occurs between two hexagons (6,6-ring junction) or a pentagon and a hexagon (5,6-ring junction), and also whether addition results in an open fulleroid or a closed fullerene structure.
Pertinently, symmetrical addition to a 6,6-junction leads to adducts, e.g. C60O, with C2v symmetry3 and unsymmetric addition to the 6,6-junction (or symmetric addition to the 5,6-junction) exhibits Cs symmetry; unsymmetric addition to a 5,6-junction reduces symmetry to C1.
At Edinburgh, we designed the azidoformate 3 in order to provide a nitrene of sufficient electrophilicity (vide infra) to bring about reaction with 'electron deficient' C60, and furthermore, to improve the solubility of the fullerene adduct by utilising the inherent lipophilicity of the 2,4,6-tri-tert-butylphenyl (supermesityl) group.
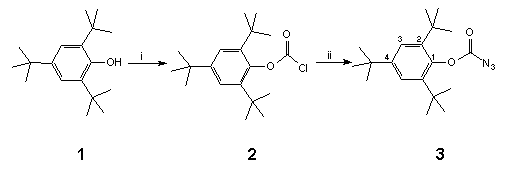
Scheme 1 Reagents and conditions: i, BunLi, 0°C, COCl2, 100%; ii, NaN3 (2 equivs.), wet acetone (0.05%), reflux, 100%.
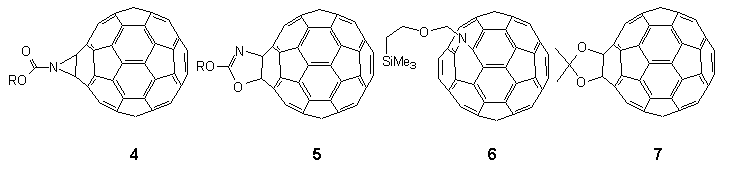
Thus, phosgenation of the lithium salt of anhydrous 2,4,6-tri-tert-butylphenol 1 led to chloroformate 2† in quantitative yield from which azidoformate 3‡ was obtained by heating with sodium azide in wet acetone overnight (Scheme 1). When a boiling solution of C60 (1 equiv.) in 1,1,2,2-tetrachloroethane (TCE) (147°C) was treated with a dilute solution of azidoformate 3 (2 equivs.) in TCE over 1 min., nitrogen evolution occurred immediately and the colour of the solution changed from purple to dark brown. The reaction mixture was heated for a further 5 min. and then poured onto crushed ice under argon to give a reaction product in 70% yield which was easily separated into two components (9:1) by preparative HPLC using a FullereneSep® RP-HPLC column10. FAB-MS analysis showed that both components were mono-adducts of C60 and 3, and formed by loss of nitrogen, the major product being characterised as the fulleroaziridine 4 (R = supermesityl) [(M+1) 1024.22767, C79H30O2N requires 1024.22764] with a closed structure on the following spectroscopic evidence. The 13C NMR spectrum (90 MHz, CDCl3/CS2) of highly soluble 4 was found to be relatively simple, consisting of four lines at 31.0, 31.4, 34.2 and 35.2 corresponding to two o-tert-butyl- and one p-tert-butyl-groups, along with five lines at 152.8, 147.1, 141.1, 123.1 and 146.2 due C=O and C1-4 of the supermesityl ligand. The region between 139.8 and 144.3 contained thirteen lines of intensity 4 and three lines of intensity 2. A further line of intensity 2 was observed at 85.6. This data suggests that the mono-adduct 4 possesses C2v symmetry which would be consistent with the incorporation of an aziridine ring at a 6,6-junction of C60 with fast pyramidal inversion at nitrogen11. The resonance at 85.6 in the sp3 region lies between those found for the cyclopropane (methanofullerene) derivative4 ( 80) and the corresponding epoxide C60O ( 90)3 and was assigned to the bridgehead sp3 carbon atoms in the fullerene cage. This finding is in sharp contrast to a report12 which appeared during the course of this work in which the heating of an equimolar mixture of [(trimethylsilyl)ethoxy]methyl azide and C60 in chlorobenzene (b.p. 132°C) overnight produced an azafulleroid 6 with an open 5,6-structure as identified from the 13C NMR resonance at 137.06 for the bridging sp2 carbon atoms. A possible explanation for this divergence in behaviour may lie in the mode of formation of 4 vis-à-vis 6. In the latter case it is proposed13 that the azafulleroid is formed via 1,3-dipolar addition and subsequent elimination of nitrogen from the resultant triazoline adduct. In our case reaction is more likely to occur via nitrene addition since analysis of the crude reaction mixture revealed the presence of other nitrene-derived products, including an isocyanate and a benzoxazolone, both of which are formed by attack on the aromatic ring of the supermesityl grouping14.
The 1H NMR spectrum of the aziridinofullerene 4 was unremarkable, consisting of three singlets at 1.55 (9H), 1.65 (18H) and 7.47 (2H), and the FT-IR (KBr) of 4 had diagnostic bands at 1752 and 526 cm¯¹. The UV-VIS spectrum of a pink-red solution of 4 in CH2Cl2 exhibited max at 226 nm ( = 3.86x104), 256 nm ( = 4.83x104) and 323 nm ( = 1.24x104) together with two weak but sharp features at 408 and 421 nm. The feature at 421 nm is reported to be highly diagnostic for closed 6,6-bridged fullerene derivatives3,9 and both these weak features are observed in the spectrum of C60O3 (vide supra). The longer wavelength spectrum between 450 and 700 nm is much less structured when compared with that of C60 and its maximum is hypsochromically shifted from 550 nm to 474 nm.
The exact molecular mass of the minor adduct [(M+1) 1024.22767] established its identity to be an isomer of 4 (vide supra). In light of our earlier studies into the thermal rearrangement of aziridine moieties15 we reasoned that this isomeric form might contain an oxazole ring such as that depicted in 5, the origin of which lies in a thermal transformation of aziridinofullerene 4 via a mechanism involving a diradical species. A control experiment confirmed that heating of a pure sample of aziridinofullerene 4 in boiling TCE over a period of 5 h resulted in the irreversible quantitative formation of 5. A kinetic study of the rearrangement under the same conditions showed the process to be first order (k = 1.3 x 10-2 s-2). Further proof of the oxazole structure came from the FT-IR (KBr) spectrum which showed that the strong C=O stretching absorption at 1753 cm-1 in 4 had been replaced by a less intense C=N absorption band at 1666 cm-1
The question remained as to the position of bridging in 5. The 13C NMR spectrum (150 MHz) of the adduct consisted of thirty-nine signals of which nine corresponded to the appended ligand which resonated at 164.1 (C=N), which we originally misassigned (vide infra) along with peaks at 31.4, 31.7, 34.5, and 35.6 (o and p-tert-butyl-groups) and 147.5,141.4, 123.4, and 147.4 (C1-4 of the supermesityl ligand). The most diagnostic of the remaining peaks ascribed to the C60 skeleton were found in the sp3 region at 97.5 (1C, br.) for the N-bound carbon and 88.6 (1C, s) for the O-bound carbon. The rest of the spectrum was observed between 135 -148 and consisted of a total of twenty-eight lines of intensity 2 and two lines of intensity 1 which would indicate Cs symmetry with the point of attachment of the supermesityl ligand across a 6,6-junction with O and N vicinally bound to the fullerene framework. The UV-VIS spectrum of 5 (yellow solution in CH2Cl2) exhibited max at 227 nm ( = 9.82x104), 255 nm ( = 1.13x105) and 316 nm ( = 4.03x104), but lacked the distinctive sharp features at 408 and 421 nm found in the spectrum observed for aziridinofullerene 4. Interestingly the spectrum of 5a in the longer wavelength region closely resembles the spectrum reported for the 1,3-dioxolane 6,6-bridged fullerene 76 formed from the reaction of C60 with 1,2-dimethyloxirane.
This series of experiments demonstrated unequivocally that decomposition of azidoformate 3a (Scheme 2, the suffix a, b or c will always refer to the groups shown in this Scheme) at 147°C in TCE in the presence of C60 produces a closed 6,6-bridged aziridino[2',3':1,2][60]fullerene 4a. We argued that this product arose by singlet nitrene addition to C60. Moreover, it was found that further heating of 4a resulted in a quantitative rearrangement to isomeric 3-(2,4,6-tri-tert butylphenyl)oxy oxazolo[4',5':1,2] [60]fullerenene 5a with both O and N vicinally bound to the fullerene skeleton at a closed 6,6-junction. Such findings exemplified the possibilities of being able to carry out chemical manipulations of functionalities on the fullerene surface.
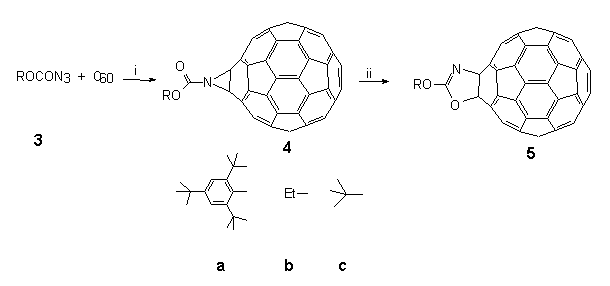
Scheme 2. Conditions: (i) TCE, 147°C, 5 min, 70%; (ii) TCE, 147°C, 12h, 100% for 4a and b only.
We next turned our attention to a much milder method for the preparation of aziridinofullerenes 4 which involved the capture of C60 by nitrenes generated at room temperature from the base-induced decomposition of substituted O-4-nitrophenylsulfonylhydroxamic acids 816,17. In light of our experience with the nitrene-mediated reaction of supermesitylazidoformate 3a to C6018 at elevated temperatures, for our initial study we synthesised O-4-nitrophenylsulfonyl-2,4,6-tri-tert-butylphenylhydroxamic acid 9a§ in a sequence of high yielding steps from supermesitylphenol (Scheme 3). Thus, treatment of the readily formed chloroformate 2a18 with hydroxylamine hydrochloride, potassium carbonate and calcium hydride in boiling DME (85°C) gave the hydroxamic acid 8a. Coupling of O-lithiated hydroxamic acid 8a with 4-nitrophenylsulfonyl chloride at -78°C in diethyl ether gave the required O-4-nitrophenylsulfonylhydroxamic acid 9a.
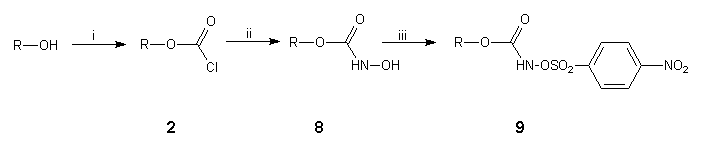
Scheme 3. Reagents and conditions: (i) n-butyl lithium, phosgene, DME, 0°C, 98%; (ii) hydroxylamine hydrochloride, potassium carbonate, calcium hydride, DME, 85°C, 94%, (iii) n-butyl lithium, n-hexane:diethyl ether, -78°C, then 4-nitrophenylsulfonyl chloride, 85%.
Treatment of a rapidly stirred mixture of C60 (1 equiv) and 9a (5 equiv) under argon in CH2Cl2/water with an aqueous solution of sodium hydrogen carbonate (5 equiv) and benzyltriethylammonium chloride (10 equiv)19 over 5 min produced an 86% yield of crude product. HPLC analysis (FullereneSep, 7% ethyl acetate:n-hexane, 2 ml.min-1, 258 nm)10 showed that 4a had been formed together with 5a in a 5:1 ratio. The formation of 5a at room temperature in this case was unexpected but can be explained by addition of the nitrene intermediate 10 in its 1,3-dipolar mesomeric form to C60 (Scheme 4).
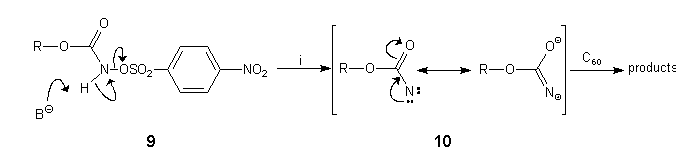
Scheme 4: Reagents and conditions: (i) sodium hydrogen carbonate, benzyltriethylammonium chloride, water/CH2Cl2, 20°C.
Following this demonstration of the applicability of the base-induced -elimination reaction as a practicable route to aziridinofullerene 4a, we turned our attention to an investigation of the effect of substituting an alkyl group in place of the supermesityl moiety. To this end we synthesised both ethyl- and tert-butyl- O-4-nitrophenylsulfonylhydroxamic acid derivatives 9b16 and 9c20 in good yield. When C60 was treated with the ethyl analogue 9b under phase-transfer conditions at room temperature, N-ethoxycarbonylaziridino[1',2':2,3][60]fullerenene 4b was formed in 65% yield and could be easily separated from unreacted C60 by flash chromatography on silica (toluene:hexane 1:10). The 13C NMR spectrum (62.5 MHz, CS2/CDCl3) of isolated 4b was as expected for a derivative of C60 with C2v symmetry and consisted of 13 lines of intensity 4 and 3 lines of intensity 2 in the sp2 region between 139.7 and 144.9, together with a diagnostic line of intensity 2 in the sp3 region at 80.5; the resonances for the ethoxycarbonyl moiety were observed at 14.5 (CH3), 64.0 (CH2) and 155.5 (C=O). The 1H NMR spectrum of 4b consisted of a triplet ( 1.60, J = 7.1 Hz) and a quartet ( 4.5, J = 7.1 Hz), whilst its FT-IR spectrum exhibited strong bands at 1743 (C=O) 1228 (C-O), and 526 cm-1. FAB-MS analysis confirmed that the product was indeed a mono-adduct [(M++1) 808.04274, C63H6NO2 requires 808.03985].
During the course of this work a report21 by Japanese workers appeared describing the synthesis of 4b by heating a mixture of C60, sodium azide, ethyl chloroformate and 15-crown-5-ether in toluene. It was claimed that the compound remained unchanged upon heating under reflux in o-dichlorobenzene (180°C) for 8 h. In our hands, it was found to rearrange to 3-ethoxyoxazolo [4',5':1,2][60]fullerenene 5b under the same conditions. An HPLC study (FullereneSep) of the rearrangement showed that at this temperature the process was accompanied by the formation of significant amounts of C60. The rearrangement was also carried out in boiling TCE (147°C) and the products were easily separated by preparative HPLC. The 13C NMR (62.5 MHz, CS2-CDCl3) spectrum of 5b showed the presence of diagnostic sp3 carbon atoms that resonated at 96.8 and 88.3, along with C=N at 164.2 (vide infra); the rest of the fullerene sp2 carbon atoms were observed between 136 and 148 (28 lines of intensity 2 and 2 lines of intensity 1). The 1H NMR (250 MHz) spectrum of the product consisted of a triplet ( 1.71, J = 7.1 Hz) and a quartet ( 4.82, J = 7.1 Hz) and its FT-IR spectrum showed characteristic absorptions at 1653 (C=N) and 526 (fullerene) cm-1. FAB-MS data confirmed this product to be an isomer of 4b [(M++1) 808.04049, C63H6NO2 requires 808.03985]. These data compare favourably with that previously reported18 for the oxazolofullerene 5a obtained from supermesitylazidoformate. The failure of the Japanese workers to detect the rearrangement of 4b may lie in the relative slowness of the process which required heating for 65 h to bring about 50% conversion at 147°C; cf. 19% conversion at 180°C for 19h, and 100% conversion in 12 h for the supermesityl analogue 4a.
In a further significant development to these findings, treatment of a solution of 4b in CH2Cl2 with a pre-mixed solution of phenol and trimethylsilyl chloride22 (3:1) in CH2Cl2 led to dealkylation and the propitious formation of the parent 1',3'-oxazolidin-2'-ono[4',5':1,2][60]fullerene 11 in 95% yield (Scheme 5). FAB-MS analysis of the product showed (M++1) at 780.00831 (C61H2NO2 requires 780.00855) and the FT-IR exhibited strong bands at 3420 (br., NH), 1760 (C=O), and 527 (fullerene) cm-1. The 13C NMR (62.5 MHz, d5-pyridine) consisted of 32 fullerene lines as expected for a derivative with Cs symmetry, the most diagnostic resonances being at 157.9 (C=O) and two lines in the sp3 region at 94.0 and 74.4; the sp2 carbon atoms of the fullerene skeleton were found between 135.2 and 147.8. It is worth noting that at this stage a report23 appeared which claimed that oxazolidin-2'-onofullerene 11 could also be prepared by treatment of 4b with boron tribromide in toluene at room temperature. The data reported for 11 was at odds with that reported by us20 (vide supra). In particular, we recorded the carbonyl group in the FT-IR of our compound to be at 1760cm-1 (cf. 1647 cm-1 for the Taiwanese group), a value that is typical; 13C NMR data was also different, and significantly, the diagnostically useful sp3 fullerene carbons were found to resonate at 94.0 and 74.4 along with the carbonyl at 157.9 (cf. 98.8, 90.5 and 166.0). Moreover, our material was found to virtually insoluble in all common solvents with the exception of pyridine whereas the Taiwanese group were able to run their 13C NMR spectrum in CS2-d6-acetone. These inconsistencies led us to re-evaluate our spectral data. It was found that the 13C NMR ( 148) shift for the C=N moiety had been misassigned and reassigned it to a broad signal at 164.2. Our data now matches closely that reported by the Taiwanese group which leads us to the conclusion that their product is probably the oxazole 5b.
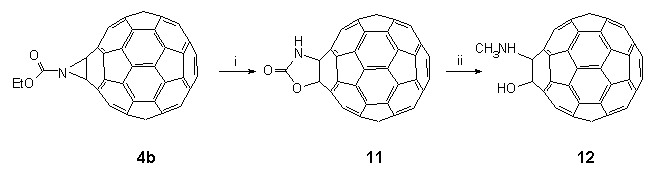
Scheme 5: Reagents and conditions: (i) phenol/chlorotrimethylsilane, (3:1), CH2Cl2, 20°C, 95%; (ii) DIBAL in CH2Cl2, DME, 20°C, 90%.
The mechanism of formation of 11 awaits further study, but in the meantime it is worth noting that its isolation highlights the opportunities of conducting reactions on functionalities attached to the surface of C60 by the following chemical transformation. Oxazolidin-2'-onofullerene 11 was suspended in DME and added dropwise to 1M DIBAL24 in CH2Cl2 at ambient temperature with stirring for 1 h. Careful addition of concentrated ammonia to the reaction mixture produced a brown precipitate in the CH2Cl2 layer which was easily isolated. FT-IR showed that the carbonyl group in the starting material (1760 cm-1 ) had disappeared and that the strongest feature in the spectrum was a broad band at 3447 cm-1. FAB-MS analysis of the product confirmed that the starting material had been reduced to C61H5NO [(M++1) 768.04371, C61H6NO requires 768.04494]. This evidence would suggest that 11 had been reduced to yield 1-hydroxy-N-methylamino[60]fullerene 12.
Developments into the functionalisation of C60 continue apace, and certain trends in the regiochemistry of addition reactions have now become apparent2,26. It has been found that all 6,6-bridged mono-adducts A (Fig. 1) of C60 possess a closed transannular bond, whilst all 5,6-bridged compounds B have an open transannular structure4,26. The rationale for these observations is that the structure of the two adducts reflect the electronic and structural preferences in the fullerene sphere, e.g. with all double bonds radiating away from pentagons and located between hexagons in a [5]radialene-like manner. There are two other structural possibilities for mono-adducts of C60; these are the 6,6-bridged compound C with an open transannular structure and the [5,6]-bridged compound with a closed transannular bond D.
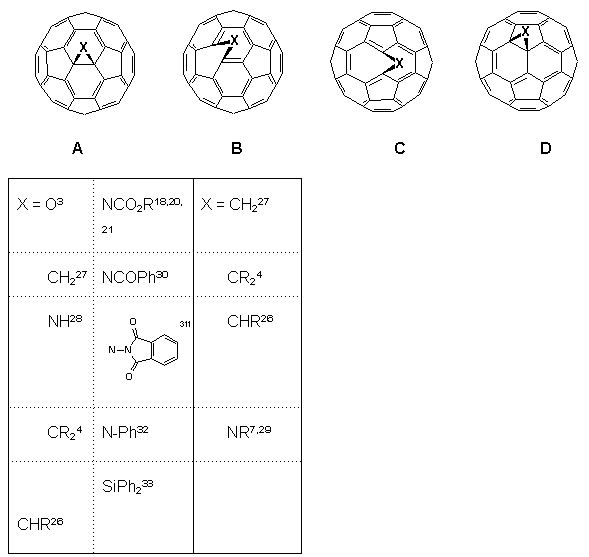
Figure 1. Four possible regioisomers arising from addition reactions of C60 (reported compounds indicated).
Hitherto, neither structure had been observed experimentally, the apparent reason being that they require the placement of double bonds in unfavorable locations, i.e. in pentagons and/or bridgeheads. Computational studies4,25,26 on the methanofullerene (X = CH2, CR2) series suggested the relative stabilities for the four isomeric structures A-D to be as follows: A (0 kJ.mol-1) > B (25 kJ.mol-1) > D (88 kJ.mol-1) with no local minima for C. This has led to the postulate that D is an intermediate in the thermal isomerisation of B (kinetic product) into A (thermodynamic product) when X = CR2, although notably it has not been possible to effect the isomerisation when X = CH227.
In the course of the studies at Edinburgh just described, HPLC analysis (Fullerenesep10, 2 ml/min, 7% ethyl acetate:n-hexane, 258 nm) of the crude reaction mixtures established that the [6,6]-bridged aziridinofullerenes 4a-c were accompanied in each case by a minor product in the ratio ca. 8:1, irrespective of the mode of formation18,20. By combining the outcome of several reaction mixtures with careful flash chromatography on silica (n-hexane:toluene), it has become possible to isolate and characterise these minor products as being isomeric with the closed [6,6]-bridged major products 4a-c. Thus, molecular ions were found by FAB-MS at the following values: 12a:(M++1) 1024.22345, C79H30NO2 requires 1024.22354; 12b:(M++1) 808.03577, C63H6NO2 requires 808.03985; 12c:(M++1) 836.06967, C65H10NO2 requires 836.07115. Further support for structural isomerism also came from the close similarity of the respective 1H NMR spectra. Comparison of the FT-IR spectra also verified the integrity of the oxycarbonylaziridine moiety with carbonyl bands at 1749.6 cm-1 for 12a (cf. 1752.3, 4a), 1731.6 for 12b (1743.0, 4b) and 1731.8 for 12c (1738.0, 4c). Likewise, the 13C NMR spectra for 12a-c showed carbonyl resonances at 152.01 ppm (cf. 152.8, 4a), 153.97 (155.5, 4b) and 153.04 (154.4, 4c) respectively, and in each case resonances due to the R group at values very close to those recorded for the 6,6-bridged aziridinofullerenes 4a-c.
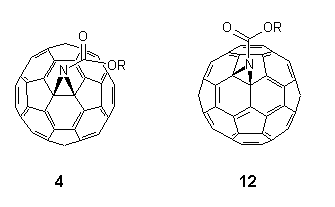
With the foregoing evidence in hand, the intriguing question remained as to the nature of the bonding in the minor isomers 12. By analysis of the 13C NMR spectroscopic data for the fullerene skeletons, in each case we are drawn to the conclusion25 that the site of substitution must be at a closed 5,6-ring junction, i.e. compounds 12 are of the hitherto unobserved structural type D (X = NCO2R). Thus, the spectra of 12 consisted of 32 lines for the fullerene carbon atoms in the expected ratio for a compound with Cs symmetry (28 lines of intensity 2 and 4 lines of intensity 1), cf. corresponding spectra of the closed 6,6-bridged major product 4 which consisted of only 17 lines for the fullerene sphere as predicted for C2v symmetry18,20,21,28. Of the 32 lines for 12, 31 were observed between 146 and 132 ppm (27 lines of intensity 2 and 4 lines of intensity 1) as well as a single and diagnostically significant line assigned to the two equivalent sp3 carbon atoms attached to the aziridine nitrogen at 104.2 ppm for 12a, (cf. 85.6, 4a), 80.4 for 12b, (80.5, 4b) and 82.6 for 12c, (80.9, 4c). In this connection, it is also worth noting that these values are distinctly different to that reported for the open 5,6-bridged compound 6 (X = NCH2OCH2CH2SiMe3) for which the sp2 carbon atoms attached to the nitrogen function resonate at 137.06 ppm12. The UV-VIS spectra (n-hexane) of minor isomers 12 all showed similar features to C60 and the closed [6,6]-bridged isomers 4 in the region between 200-400 nm with absorbances occurring at max ca. 210 nm ( = 5x104 dm3mol-1cm-1), 256 (4x104) and 325 (9x103). However, they lacked the distinctive fine structure observed in C60 and closed [6,6]-bridged compounds in the region ca. 405-425 nm3,9,18,20,28,34. In addition, relative to the closed 6,6-bridged compounds 4 the closed 5,6-isomers 12 showed a slight red shift (3-5 nm).
At this stage, little is known about the mechanistic origin of the minor closed 5,6-isomers 12. The formation of the major closed 6,6-isomers can be rationalised in terms of trapping by C60 of a singlet nitrene and it is proposed that the minor isomeric products 12 may result from intersystem cross-over with the nitrene acting in a triplet diradical manner (Fig. 2). Thus, after the the initial radical addition reaction has occurred at position 1, the unpaired electron is mostly located on two fused hexagons with the highest spin densities at positions 2, 4 and 635. Radical recombination at position 4 seems unlikely, whereas recombination at position 2 gives rise to the closed 6,6-isomers 4; recombination at position 6 would lead to the observed formation of closed 5,6-isomers 12.
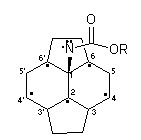
Figure 2. Part structure of C60 denoting delocalised position of unpaired electron following addition of triplet nitrene, ROCON:
Further chromatography afforded another fraction that was shown by a combination of MALDI and FAB-MS analysis to contain bis-adducts, which according to 13C NMR spectroscopy contained at least five isomeric forms. For example, five different C=O resonances were observed at ca. 153 along with several resonances at 80 (sp3). In the sp2 region, the mixture exhibited at least 200 lines !
Compared to the 6,6-bridged aziridinofullerenes 4a,b, heating of the tert-butoxycarbonyl derivative 4c follows a separate pathway with simultaneous loss of isobutene and CO2 to form aziridino[2',3':1,2][60]fullerene (C60NH) 13, which in sharp contrast to C60O is a thermally stable derivative which can be heated in boiling TCE (147C) without change. Thus, the synthesis of bronze-coloured 13 is conveniently achieved by a two-step process as outlined in Scheme 6.
The first step involved dropwise addition to a solution of C60 in boiling TCE of tert-butylazidoformate 3c36, which is synthesised and stored as a 3M stock solution in TCE thus minimising the intrinsic danger of azides37. The outcome was elimination of nitrogen, and within minutes, the formation of N-tert-butoxycarbonylaziridino[2',3':1,2][60]fullerene 4c¶ , by in situ trapping of the intermediate nitrene (tert-BuO2CN:) with C6018. The same result could also be achieved, albeit under much milder conditions, by base-induced -elimination20 of O-4-nitrophenylsulfonyl-tert-butylhydroxamic acid 9cII in the presence of C60 under phase-transfer conditions at room temperature19. By both these procedures, compound 4c is obtained in 55-60% yield, and easily purified by flash chromatography on silica (n-hexane-toluene).
In the second step, compound 4c was heated in TCE (5 h), whence elimination of isobutene and CO2 occurred to form the key compound, C60NH 13, in 70% yield28. No evidence was found for a fullerenoaziridine-oxazole (or aziridinofullerene-oxazolofullerene) rearrangement previously observed for this class of compounds18,20,30,37. Pertinently, no molecular ion at 835 (C65H9NO2 requires 835) was observed in the FAB mass spectrum of 4c, but ions at 780.00969 [(M++1) C61H2NO2 requires 780.00855] and 736.01497 [(M++1) C60H2N requires 736.01872] were measured and correspond to the sequential loss of isobutene and CO2 from 4c.
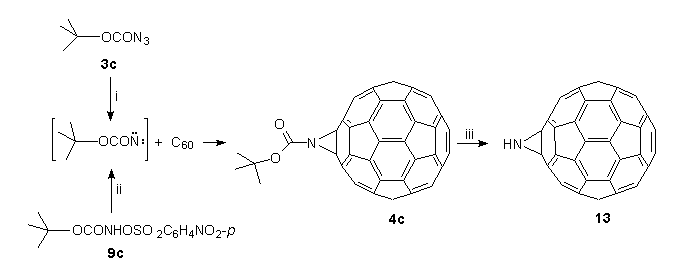
Scheme 6. Reagents and conditions: (i) TCE, 147C; (ii) NaHCO3, BzEt3NCl, H2O/TCE/DCM, 20;(iii) TCE, 147C, 5 h.
The structural assignment of 13 rests on spectroscopic arguments, and in the interim, attempts are being made to produce a highly crystalline derivative for X-ray crystallographic analysis. FAB-MS analysis 13 showed a prominent molecular ion at 736 [(M++1) 736.01199, C60NH2 requires 736.01872]. The 13C NMR spectrum is consistent for a molecule with C2v symmetry incorporating an aziridine ring at a 6,6-junction of C60 with fast pyramidal inversion at nitrogen11. Hence, there are fifteen lines in the fullerene region 147 and 138 ppm (1 line of intensity 8, 11 lines of intensity 4 and 3 lines of intensity 2) and a diagnostically significant peak in the sp3 region at 79.12 ppm (CS2-2[H6]acetone) [78.71 ppm (CS2-CDCl3)]* ; lines due to the carbonyl and tert-butyl carbon atoms in the precursor 4c were absent. Similarly, in the 1H NMR spectrum of 13 the resonance at 1.7 ppm due to the tert-butyl group had disappeared and replaced by a broad signal assignable to NH at 5.9 ppm, which disappeared on deuteriation. In the FT-IR (KBr) spectrum of 13 there was no carbonyl band and the main bands were 3272 (w, NH), 1426.6, 1184.0, 1039.8, 706.3, 615.7, 566.7, 526.3 and 497.1 cm-1. It is noteworthy that four of these band resemble the principal absorptions of C60 (1429.0, 1182.7, 575.9, 526.9)3. The UV-VIS spectrum of the faintly pink CH2Cl2 solution of 13 displayed typical absorptions due to the fullerene skeleton at max 258.5 nm( = 3.05x104 dm-3mol-1cm-1), and 326.5 (9.03x103), together with a shoulder at 410.5 and a weak but sharp feature at 423.5 (1.94x103). These data compare favourably with those reported for closed 6,6-compounds such as C60O3 (1,2-epoxy[60]fullerene) and C61H227 (1,2-methano[60]fullerene) and so it is concluded that the structure of 13 is that of a closed [6,6]-aziridinofullerene.
In passing it is worth noting that thermolysis of closed 5,6-bridged adduct 12c in TCE under reflux for 8h resulted in elimination and isomerisation to produce closed 6,6-bridged 13. Furthermore, thermolysis of the isomeric mixture of bis-adducts derived from tert-butoxycarbonylnitrene produced a complex mixture of isomeric bis-aziridinofullerenes C60N2H2. An interesting feature of the thermal degradation is the appearance of minor amounts of closed 6,6-mono-adducts 4c together with its closed 5,6-isomer 12c in addition to C60NH 13.
We are currently exploring the potential of 13 to undergo ring-opening reactions of strained aziridines, thus providing a valuable route to 1,2 addition products and their further elaboration. We are also investigating the functionalisation of 13, and in this connection it is worth mentioning the quantitative formation of D-galactose derivative 14** by direct acylation under mild conditions (Scheme 7).
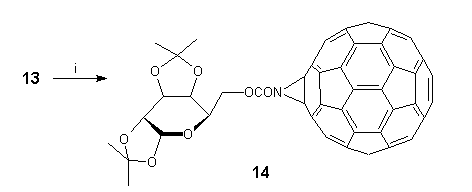
Scheme 7. Reagents and conditions: (i) 1,2:3,4-di-O-isopropylidene-D-galactopyranose-6-chloroformate, pyridine, TCE, 22C, 2h.
Compared to the plethora of methods that now exist for the mono-functionalisation of C60 via thermal cycloaddition reactions2, photochemical derivatisation of C60 is still in its infancy. Reports so far include photo-epoxidation3, [2+1]-photocycloaddition reaction with nitrenes derived from N-succinimidyl-4-azido-2,3,5,6-tetrafluorobenzoate32 and aroylazides30, [2+2]-photocycloaddition reaction with enones38 and N,N-diethylpropynylamine5, [2+3]-photocycloaddition reactions with 2,3-diphenyl-2H-azirine39 and 1,1,2,2-tetramesityl-1,2-disilirane33. In all cases, functionalisation occurs at a 6,6-ring junction in C60, which in the photoexcited state is a much stronger electron acceptor and serves as an ene or dienophile. We have already established that in addition to the expected formation of closed 6,6-fulleroaziridines 4 by reaction of C60 with singlet oxycarbonylnitrenes18,20, e.g. EtO2CN:, small amounts (ca. 10%) of unanticipated closed 5,6-adducts 12 were obtained, apparently by trapping of triplet nitrenes in a diradical manner25. In an effort to investigate this conjecture further, photolytic experiments were undertaken in various solvents, and in particular benzene with divergent results40. Thus, irradiation (400 W medium pressure mercury lamp, quartz reactor) of a mixture of C60 (1 equiv.) and 3b (5 equiv.) in either CH2Cl2 or TCE led to the immediate formation of an intractable brown solid, whereas in benzene, slow consumption of C60 occurred to produce a 4:1 mixture of two major products (70 %), which was easily separated by flash chromatography (n-hexane:toluene, n-hexane:ether). Analysis of the crude reaction mixture by HPLC (FullereneSep, 2 ml/min, 7% ethyl acetate:n-hexane, 258 nm)10 also established the concomitant formation of small amounts of closed 6,6-bridged aziridinofullerene 4b (3%); when the C60:ethyl azidoformate ratio was changed to 1:60, closed 5,6-bridged aziridinofullerene 12b (ca. 1%) was also detected. FAB-MS analysis showed that the two products were isomeric [major (M++1) 886.08650, minor (M++1) 886.07837; C69H12NO2 requires 886.08680] and corresponded to an adduct of C60 combined with benzene and photolytically-generated ethoxycarbonylnitrene. Control experiments established that reaction had proceeded by capture of the nitrene by benzene to produce N-ethoxycarbonylazepine 15, which then underwent further photoreaction with C60 by two competing pathways to form the isomeric adducts. The photogenesis of these adducts was established when azepine 15 was found to be thermally unreactive towards C60 upon heating in benzene under reflux for 3 h in the dark. By contrast, photolysis of 15 in the presence of C60 in benzene produced the same reaction mixture as obtained in the initial experiment.
The structures of the major and minor photoadducts were elucidated by a series of high-field NMR experiments, which established unequivocally that their origin resulted from formal [2+4]- and [2+6]-cycloaddition reactions of 15 to C60, respectively as depicted in Scheme 8. In the case of the major [2+4]-photoadduct 16, the one-dimensional 1H NMR spectrum (600 MHz) showed six pairs of signals in the 4-8 ppm region as follows: overlapping triplets at 4.21 ppm, triplets at 5.51 (major) and 5.64 ppm, doublets at 6.52 and 6.68 (major) ppm, overlapping triplets at 6.90 ppm, doublets at 7.25 (major) and 7.35 ppm, overlapping triplets at 7.50 ppm. In each case saturation of one of the members of a resolved pair resulted in saturation transfer to the other, indicating exchange between two conformers or isomers. A two-dimensional HMQC41 carbon-proton correlation experiment showed that the protons resonating at 4.21 (two), 6.52 and 6.68 ppm are bonded to sp3-hybridised carbon atoms at 44.7/44.9, 58.2 and 58.8 ppm, respectively; the remaining protons being bonded to alkene carbon atoms. Proton spin-decoupling experiments confirmed that the protons constitute a linear spin system in which two alkene units are separated by an sp3 hybridised carbon atom. Above 60C the pairs of proton signals coalesced to show sharp doublets or triplets as expected. The 13C NMR spectrum (62.5 MHz) also exhibited signals at 71.5 and 69.7 ppm, which were assigned to sp3 carbon atoms of the fullerene sphere, along with 35 (out of 60) resolved lines between 135 to 155 ppm for the fullerene sphere. Resonances due to the alkene portion of 16 were observed at 139.0, 127.0, 124.5 and 108.7 ppm, whilst the N-ethoxycarbonyl moiety had resonances at 14.3, 62.6 and 153.7 ppm; both of these sets of signals occurred as major/minor pairs. The FT-IR spectrum.(KBr) exhibited bands at 1704 (C=O), 1647 (C=C), 1323, 1254, 725 and 526 (fullerene) cm-1. The UV-VIS spectrum (DCM) of 16 exhibited a maximum at 309 accompanied by shoulders at 415 and 435 nm.
E = COOR (R = Et, But)
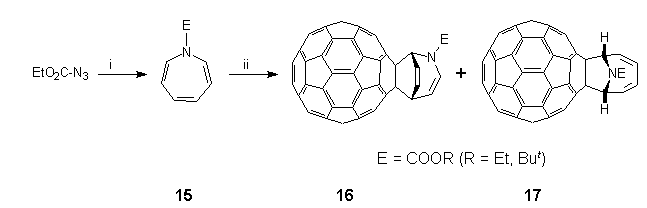
Scheme 8. Reagents and conditions: (i) benzene, h; (ii) C60.
For the minor [2+6]-photoadduct 17, more elaborate NMR experiments were required to determine its structure. Thus, in addition to the methyl (1.48 ppm), and diastereotopic methylene (4.41 and 4.44 ppm) resonances, the one-dimensional 1H NMR spectrum (600 MHz) showed a two-proton multiplet centred at 6.31 ppm, similar in appearance to one portion of an [AX]2 spectrum, a one-proton doublet at 6.36 ppm showing a further long-range coupling, and a complex pattern, integrating for three protons, from 6.40-6.48 ppm. A saturation difference experiment with irradiation at 6.36 ppm revealed a similar doublet at 6.43 ppm. A two-dimensional HMQC41 carbon-proton correlation experiment showed that these two protons are bonded to sp3 hybridised carbon atoms at 67.5 and 67.7 ppm respectively, the remaining four protons being bonded to alkene carbon atoms at 137.1, 136.6, 125.4 and 125.1 ppm. Examination of the two-dimensional proton DQFCOSY42 spectrum located the remaining two protons at 6.42 and 6.46 ppm and confirmed the shift of the other four protons. The three-bond coupling cross peaks showed that the protons constitute a linear spin system which terminates with protons bonded to sp3 hybridised carbon atoms. The exchange of the terminal protons arises from the rotation about the N-CO2Et bond which is slow on the NMR time-scale at the temperature (25C) at which the spectra were obtained. Hence the symmetry expected in a fast exchange situation, which occurs at a temperature higher than could be made on this sample, is not observed. Owing to its Ci -symmetry, the 13C NMR spectrum (62.5 MHz) of photadduct 17 showed two fullerene carbon sp3 resonances at 81.5 and 80.4 ppm, along with 43 (of 60) resolved peaks between 134.2 to 153.6 ppm; the N-ethoxycarbonyl moiety also showed resonances at 14.6, 61.91 and 153.1 ppm. FT-IR spectrum (KBr) displayed major absorbances at 1705 (C=O), 1509, 1316, 1262, 717 and 525 (fullerene) cm-1. The UV-VIS spectrum (DCM) of 17 showed maxima at 303 and 426 nm. The sharp but weak feature at 426 nm is characteristic of 6,6-substituted fullerenes3,5,18,20,30,33.
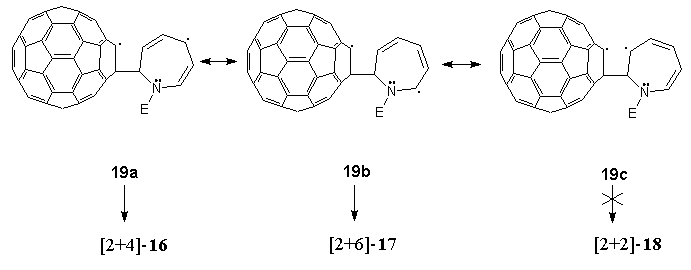
From a mechanistic viewpoint, the previously unobserved modes of photo-induced addition of azepine 15 to C60 can be accounted for in terms of a photoexcited (triplet) state43 of the latter forming a metastable exciplex with electron-rich 15. Subsequent decay to diradical intermediates such as 19a-c can be visualised in which delocalisation as indicated accounts for the preferred formation of photoadduct 16 over photoadduct 17. We failed to detect the formation of [2+2]-photoadduct 18, presumably due to a combination of two factors. Firstly, of the three diradical intermediates 18a-c proposed, the one that would result in a [2+2]-photoadduct, i.e. 19c, is the least stable, and secondly, formation of a four-membered ring is energetically less favourable than the larger rings formed in the observed alternative products, 16 and 17.
References