 ,
, -unsaturated acids gave substrates for Diels-Alder
reactions [1] and Michael additions [2] in which some excellent stereoselectivities
and yields were attained.
-unsaturated acids gave substrates for Diels-Alder
reactions [1] and Michael additions [2] in which some excellent stereoselectivities
and yields were attained.
1
Figure 1
We were also interested in the application of these compounds as potential modifiers or ligands for asymmetric synthesis, a subject which has been the focus of intense interest in recent years.[3] The use of an additive which can induce asymmetry into a process and be recovered unchanged after the completion of the reaction is appealing, particularly if this can be carried out catalytically. However, the hydroxyl groups in 1 are clearly not correctly disposed for ligation of a single metal atom. To solve this problem we envisaged a fundamental alteration to the dispiroketal framework. Models suggested that the use of bis-dihydrofuran-derived diols 3 (Scheme 1) would give a complex in which the hydroxyl groups project into the cleft formed by the two furanyl rings, holding the metal close to the ligand thus enhancing asymmetric induction.
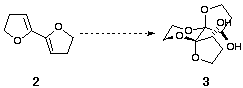
Scheme 1
Rather than starting with the achiral bis-dihydrofuran 2, which would necessitate the subsequent manipulation and resolution of the diols, we decided to use optically pure, substituted bis-dihydrofurans such as 4 or 5 (Figure 2). It was hoped that the substituents would exert control over the geometry of the anomeric centres formed during dispiroketalisation as is true in the case of dihydropyranyl dispiroketals.[4] However, prediction of the preferred orientation of the substituents on the furan rings and hence the geometry of the anomeric centres is more complicated than in the case of the dihydropyranyl dispiroketals since all the substituents are able to adopt pseudo-equatorial orientations.
In this paper we describe the synthesis of two new dihydroxylated dihydrofuranyl systems derived from dienes 4 and 5.
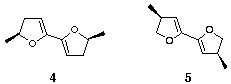
Figure 2
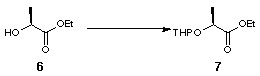
The novel 2,2'-substituted bis-dihydrofuran was synthesised starting from (S)-ethyl lactate, 6 (Scheme 2). Tetrahydropyranyl protection of the free alcohol gave 7 with a 1.2:1 mixture of epimers at the anomeric centre. DIBAL reduction of the resulting tetrahydropyranyl ester gave aldehyde 8 which was converted to olefin 9 by Wittig olefination. Hydrogenation using Adams' catalyst gave 10 and subsequent deprotection using acidic methanol followed by lactonisation furnished 11 (Scheme 2).
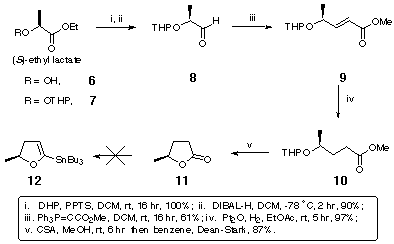
Scheme 2
We had envisaged a palladium-catalysed coupling of the vinyl stannane 12 formed from the lactone 11. We had hoped to achieve this following methodology previously established within the group during work towards the synthesis of enantiopure substituted bis-dihydropyrans.[5] This involves transformation of the appropriate lactone of the type 13 into the corresponding anomeric sulfone 15 via the lactol 14 (Scheme 3). Quenching of the sulfone anion with tributyltin chloride gives the intermediate stannylated sulfone 16 which, under mildly basic conditions, eliminates benzenesulfinic acid to furnish the vinyl stannane 17.
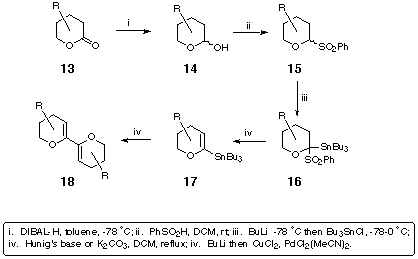
Scheme 3
When this strategy was applied to our five-membered ring lactone 11, formation of sulfone 19 proceeded smoothly (Scheme 4), but attempts to form the anion and quench with tributyltin chloride were unsuccessful (Scheme 4).
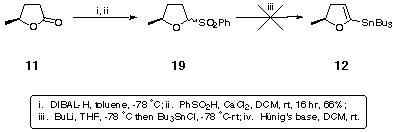
Scheme 4
This was because anion 21 was prone to decomposition, possibly by a mechanism similar to the decomposition of tetrahydrofuranyl anion 20 (Scheme 5).
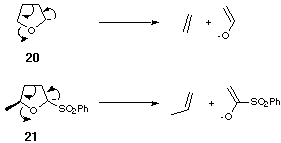
Scheme 5
This result forced us to abandon the use of vinyl stannane methodology in favour of more traditional dihydrofuran anion formation. Benzenesulfinic acid was eliminated from the anomeric sulfone 19 to give the dihydrofuran 22 using methodology developed within our group.[6] The volatility of 22 necessitated its isolation by distillation and its use as a 4% solution in THF. Anion formation and application of the coupling conditions established for bis-dihydropyran[7] gave the desired (S,S) dimethyl bis-dihydrofuran 4 (Scheme 6).
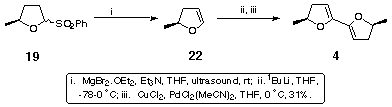
Scheme 6
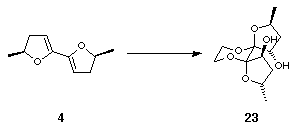
Pleasingly, dimethyldioxirane epoxidation of 4 followed by trapping with ethylene glycol under thermodynamic, acidic conditions resulted in the exclusive formation of diol 23 as a single diastereomer (Scheme 7).
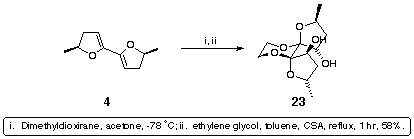
Scheme 7
X-ray structure determination revealed that epoxidation had proceeded anti to the methyl substituents; the orientation of these methyl groups was endo and the hydroxyls were exo (Figure 3).
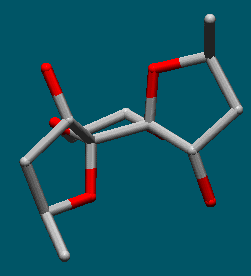
Figure 3
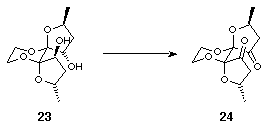
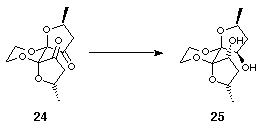
The orientation of the hydroxyls in 23 was clearly unsuitable for participation in bidentate chelate complex formation, and their inversion was therefore attempted. An activation/inversion strategy was discounted since the orientation of the furan rings would hinder the approach of the nucleophile on the required trajectory for an SN2 displacement. An oxidation-reduction strategy was thought to be more viable since it was envisaged that reduction of the diketone would occur from the open face of the furan rings to furnish the desired diol. Indeed, Swern oxidation of 23 to the diketone 24 proceeded in 71% yield and subsequent lithium aluminium hydride reduction gave exclusively the all endo diol 25 in 88% yield (Scheme 8).
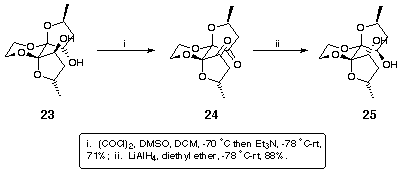
Scheme 8
When more substantial quantities of 25 become available we envisage trying to emulate Noyori's binapthol, a chiral diol which has been used as a chelating ligand with much success; for example, it is used modify lithium aluminium hydride, giving BINAL-H 26 and 27 (Figure 4), which achieves efficient asymmetric reduction of ketones bearing unsaturated substituents. [8]
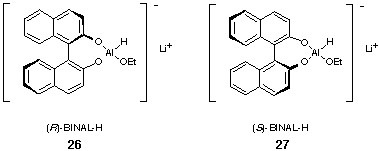
Figure 4
The second class of ligands required the preparation of the 4,4'-dimethyl-substituted bis-dihydrofuran 5. This was synthesised from the commercially available butyrolactone 28 using the anomeric sulfone methodology that had already been established for the synthesis of 4. Thus DIBAL-H reduction of 28 followed by treatment with benzenesulfinic acid furnished the anomeric sulfone 29. Elimination, vinyl anion formation and application of standard bis-dihydrofuran coupling conditions[7] afforded the desired (S,S) dimethyl bis-dihydrofuran 5 (Scheme 9).
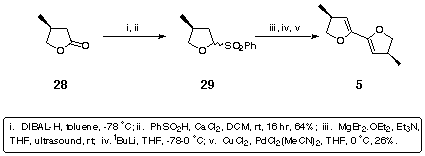
Scheme 9
Epoxidation of 5 followed by trapping with ethylene glycol could have given either or both of two possible dihydroxylated dispiroketals, 30 and 31 (Scheme 10).
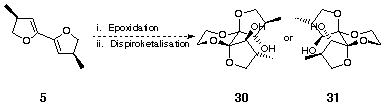
Scheme 10
In fact, treatment of 5 with dimethyl dioxirane followed by trapping with ethylene glycol under themodynamic, acidic conditions resulted in the exclusive formation of a single symmetrical diol which was shown to be 30 (Scheme 11).
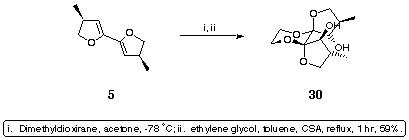
Scheme 11
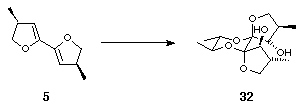
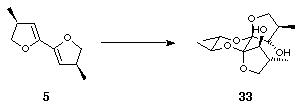
The product was shown to be 30 rather than 31 as follows. As referred to earlier, optically pure diols may be used during the thermodynamic dispiroketalisation step to control the geometry of the newly formed anomeric centres4. Thus, bis-dihydrofuran 5 was treated with dimethyldioxirane and then under standard dispiroketalisation conditions with (2S,3S)-2,3-butanediol. A sole diol product 32 was formed (Scheme 12) because the absolute stereochemistry of the spiro centres, (S,S), in the new diol was controlled by a combination of multiple anomeric effects and the absolute configuration at the site of substitution of the methyl groups on the butane diol, which adopted equatorial orientations, giving 32. Likewise, with (2R,3R)-2,3-butanediol the absolute stereochemistry of the spiro centres in the product was (R,R) and therefore a single diol 33 was formed (Scheme 12).
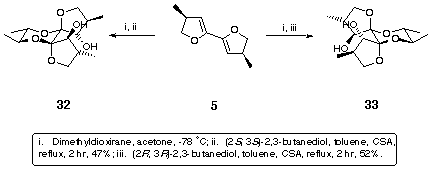
Scheme 12
The NMR data for the furanyl ring protons and carbons in 33 bore very close relation to the NMR data for 30 whereas the data for 32 and 30 were dissimilar. Although not conclusive, these results allowed us to postulate with a high degree of certainty that the product of the dispiroketalisation of 5 with ethylene glycol was 30 rather than 31. Inversion of the two alcohols is therefore necessary to make bidentate chelation possible; even then, the syn methyl substituents may hinder complexation. Diol 33, in which the enantiopure diol has forced the methyl substituents exo, is therefore a better candidate for effective ligation.
In conclusion, we have been able to synthesise two novel diols 25 and 33 which may have possible applications as chiral modifiers and ligands for asymmetric synthesis. Work is ongoing in our laboratories to investigate their efficiency at inducing asymmetry in a variety of processes and to develop more efficient routes to substituted bis-dihydrofurans.
We gratefully acknowledge the S. E. R. C. and the E.P. S. R. C. for funding (to G. H. C.) and Glaxo Research and Development for a Studentship (to J. E. I.). Further financial support was provided by Zeneca and the BP endowment (to S. V. L.) at Cambridge. We thank Dr. Jonathan Goodman and Dr. Keith Trollope, of Cambridge University, for kind assistance in preparing this paper in electronic format.
1. B. C. B. Bezuidenhoudt, G. H. Castle, J. V. Geden, S. V. Ley,
Tetrahedron Lett., 35, 7451 (1994).
2. G. H. Castle, S. V. Ley, Tetrahedron Lett., 35, 7455
(1994).
3. Monographs: (a) Asymmetric Synthesis, Vol. 5, J. D. Morrison, Ed.,
Academic Press, New York, 1985;
(b) B. Bosnich, Asymmetric Catalysis, Martinus Nijhoff, Dordrecht,
1986;
(c) Catalytic Asymmetric Synthesis, I. Ojima, Ed., VCH, Weinheim, 1993;
(d) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley
Interscience, New York, 1993.
4. G.-J. Boons, D. A. Entwistle, S.V. Ley, M.Woods, Tetrahedron Lett.,
34, 5649 (1993).
5. P. J. Edwards, D. A. Entwistle, S.V. Ley, D. R. Owen, E. J. Perry,
Tetrahedron: Asymmetry, 5, 553 (1994).
6. D. S. Brown, Ph.D. Thesis, University of London, 1990.
7. S. V. Ley, R. Leslie, P. D. Tiffin, M.Woods, Tetrahedron Lett.,
33, 4767 (1992).
8. (a) R. Noyori, I. Tomino, Y. Tanimoto, J. Am. Chem. Soc., 101,
3129 (1979);
(b) R. Noyori, I. Tomino, M. Nishizawa, J. Am. Chem. Soc., 101,
5843 (1979);
(c) R. Noyori, I. Tomino, Y. Tanimoto, M. Nishizawa, J. Am. Chem. Soc.,
106, 6709 (1984);
(d) R. Noyori, I. Tomino, M. Yamada, M. Nishizawa, J. Am. Chem. Soc.,
106, 6717 (1984).
(e) R. Noyori, Pure Appl. Chem., 53, 2315 (1981).
9. Dictionary of Organic Compounds, 5th Edition, Chapman and Hall, New
York, 1982.
Preparation of (2S) Ethyl 2-(tetrahydro-2H-pyran-2-yloxy)
propanoate 7.
(S)-Ethyl lactate 6 (25.0 g, 217 mmol) and dihydropyran
(30 ml) were dissolved in DCM (250 ml) and the resulting colourless solution
was treated with PPTS (2.5 g) at 0 C. The solution was stirred under
argon for 24 hours and then quenched by the addition of saturated sodium
bicarbonate solution (200 ml) and stirred vigorously for 10 minutes. The
organic layer was then separated, dried (MgSO4), and the solvent removed in
vacuo to yield the tetrahydropyranyl ether 7 (44.0 g, 217
mmol, 100 %) as 1.2:1 mixture of anomers, as a colourless oil;
Preparation of (2S) 2-(tetrahydro-2H-pyran-2-yloxy)propanal
8.
The THP ether 7 (45.0 g, 222 mmol) was dissolved in DCM (400 ml)
and cooled to -78 C under argon before dropwise addition of DIBAL-H
(1.5 M in toluene, 148 ml, 222 mmol) over 30 minutes. The reaction was
stirred at -78 C for 90 minutes before quenching by the portionwise
addition of sodium sulphate decahydrate (20 g) and the suspension was allowed
to come to room temperature. The solid was filtered off and was washed with
DCM (200 ml). The combined filtrates were dried (MgSO4) and then concentrated
in vacuo to yield the aldehyde 8 (31.7 g, 200 mmol, 90 %)
as a 1.2:1 mixture of anomers as colourless oil;
Preparation of (4S,2E) Methyl
4-(tetrahydro-2H-pyran-2-yloxy)pent-2-enoate 9.
The aldehyde 8 (30.7 g, 200 mmol) was dissolved in DCM (400 ml)
and to the resulting colourless solution at room temperature was added
methyl(triphenyl-phosphoranylidene)acetate (200 g, 600 mmol, 3 eq.) in one
portion. The resulting colourless solution was stirred at room temperature
under argon for 16 hours before concentration in vacuo. The resulting
pink solid was triturated with 50% ether in petrol (1 litre), filtered and
the filtrate concentrated in vacuo before purification by flash column
on silica gel (150 g in a 9 cm diameter column) with 50% ether in petrol as
eluent to yield the ester as a 1.4:1 mixture of anomers 9
(26.2g, 122mmol, 61%) as a colourless oil;
Preparation of (4S) Methyl
4-(tetrahydro-2H-pyran-2-yloxy)pentanoate 10.
The THP ether 9 (24 g, 112 mmol) was dissolved in ethyl acetate
(300 ml). The resulting solution was degassed and placed under argon. Adam's
catalyst (1.0 g) was added and the solution was degassed then placed under an
atmosphere of hydrogen. After stirring vigourously for 5 hours the reaction
was again degassed and placed under a argon atmosphere. The solution was
filtered through a pad of Celitereg. and the filtrate concentrated
in vacuo to yield the ester 10 (23.5 g, 109 mmol, 97%) as
13:1 mixture of anomers as a colourless oil;
Preparation of (5S) 5-Methyl-2(3H)-furanone 11.
The THP ether 10 (23 g, 108 mmol) and camphorsulphonic acid (1 g)
were dissolved in methanol (300 ml) and stirred at room temperature under argon
for 6 hours. The methanol was then removed in vacuo and replaced with
benzene (300 ml). The flask was fitted with a distillation apparatus. Benzene
(150 ml) was removed by distillation and the reaction allowed to come to room
temperature before quenching with saturated sodium bicarbonate solution (300
ml). The organic layer was separated and the aqueous layer was then extracted
with DCM (2 x 200 ml). The combined organic layers were dried (MgSO4), and the
solvent removed in vacuo to yield a pale yellow oil. This crude product
was purified by flash column chromatography on silica gel (200 g in a 4 cm
diameter column) with 20% ether in petrol as eluent to yield the lactone
11 (9.4 g, 94 mmol, 87%) as a colourless oil; [
Preparation of (2S/R,5S)
2-Benzenesulfonyl-5-methyl-tetrahydrofuran 19.
To a solution of the lactone 11 (9.4 g, 94 mmol) in THF (250 ml) at -78
C under argon was added DIBAL-H (1.5 M in toluene, 68.8 ml, 103 mmol,
1.1 eq.) dropwise over 15 minutes so as to maintain the temperature below
-60 C. The resulting colourless solution was stirred at -78 C
for 3 hours. The reaction was quenched by the addition of sodium sulfate
decahydrate (10 g) over 20 mins. The resulting suspension was allowed to come
to room temperature, stirred for 3 hours, filtered washing with ethyl acetate
(300 ml) and the filtrate concentrated in vacuo to yield the
lactol as a colurless oil (unweighed).
To a solution of this crude lactol and calcium chloride (35.8 g, 282 mmol, 3
eq.) in DCM (200 ml) at room temperature under argon was added, in one portion,
benzene sulphinic acid (26.6 g, 187 mmol, 2 eq.). The resulting white
suspension was stirred at room temperature for 16 hours. The reaction was
filtered and the residues washed with DCM (300 ml). The filtrate was washed
with saturated sodium bicarbonate solution (2 x 150 ml), dried (MgSO4), and the
solvent removed in vacuo to yield a pale yellow oil. This crude product
was purified by flash column chromatography on silica gel (300 g in a 9 cm
diameter column) with 50% ether in petrol as eluent to yield the anomeric
sulfone 19 as a 2:1 mixture of anomers (14.0 g, 62.0
mmol, 66%) as a colourless oil;
Preparation of (2S,2'S)
2,2'-dimethyl-2,2',3,3'-tetrahydro-5,5'-bi-furan 4.
The anomeric sulphone 19 (5.0 g, 22 mmol) in THF (15 ml) was
added to a solution of magnesium bromide etherate (11.41 g, 44.2 mmol, 2 eq.)
and triethylamine (4.62 ml, 3.35 g, 33.1 mmol, 1.5 eq.) in THF (35 ml)
under argon. The heterogeneous mixture was sonicated for 2 hours after which
time no starting material remained by TLC. The flask was then fitted with a
distillation apparatus and all the volatile components of the reaction mixture
distilled over. The distillate was then cooled to -78 C under argon
and treated with tert-butyllithium (1.7 M in pentanes) (12.1 ml, 20.7
mmol, 1.1 eq). The resulting pale yellow solution was allowed to come to 0
C and stirred for 30 minutes. It was then cooled down to -78 C
and transferred by cannula to a solution of copper (II) chloride (2.78 g,
20.7 mmol, 1.1 eq) and palladium (II) chloride bisacetonitrile
complex (110 mg, 4.2 x 10-4 mol, 2.25 mol%) in THF (20 ml) at
0C. The resulting black solution was stirred for 1 hour at 0 C
and then quenched by the addition of 0.88 ammonia solution (6 ml) in saturated
ammonium chloride (24 ml). The resulting solution was stirred vigorously at
room temperature for 30 minutes to ensure the total oxidation of copper (I)
salts. The reaction mixture was then filtered through a pad of
Celitereg. washing with ether (20 ml). The aqueous layer was
extracted with ether (3 x 40 ml), the combined organic layers were dried
(MgSO4), and the solvent removed in vacuo to yield a brown oil. This
crude product was purified by flash column chromatography on silica gel (20 g
in a 1 cm diameter column) with 2% ether in petrol as eluent to yield the
diene 4 (506 mg, 3.42 mmol, 31%) as a colourless oil; [
Preparation of
(2S,4S,5R,6R,8S,10S)
2,8-dimethyl-1,7,11,14-tetraoxa dispiro[4.0.4.4]tetradecan-4,10-diol 23.
To an acetone solution of dimethyl dioxirane (c.a. 0.05 M, 250 ml) at
-78 C was added a solution of the bis-dihydrofuran 4
(260 mg, 1.56 mmol) in ether (1 ml). The solution was stirred at this
temperature for 2 hours before being allowed to come to room temperature. The
solvent was then removed in vacuo and the resulting white solid
suspended in toluene (5 ml). Ethylene glycol (5 ml) and CSA (50 mg, cat.) were
then added and the solution heated at 120 C for 2 hours with vigorous
stirring. The resulting brown solution was allowed to come to room temperature
and was poured into saturated sodium bicarbonate solution (20 ml). The mixture
was stirred for 5 minutes and DCM (100 ml) added. The organic layer was
separated and the aqueous layer extracted with DCM (3 x 100 ml). The combined
organic phases were dried (MgSO4), and the solvent removed in vacuo to
yield a yellow oil. This crude product was purified by flash column
chromatography on silica gel (40 g in a 2 cm diameter column) with ether as
eluent to yield the diol 23 (234 mg, 8.99 x 10-4
mol, 58%) as colourless prisms; m.p. 167 C (ether/petrol);
[
Preparation of (2S,5R,6R,8S)
2,8-dimethyl-1,7,11,14-tetraoxadispiro [4.0.4.4]tetradecan-4,10-dione 24.
Oxalyl chloride (1.07 ml, 1.56 g, 12.3 mmol, 5 eq.) was dissolved in dry
DCM (10 ml) under argon and cooled to -78 C before the dropwise
addition of DMSO (1.05 ml, 1.15 g, 14.7 mmol, 6eq.) in DCM (2.5 ml) over 10
minutes so that the reaction temperature was maintained below -60 C.
Once the evolution of carbon monoxide had ceased the reaction mixture was
stirred for a further 15 minutes before the dropwise addition of the diol 23
(640 mg, 2.46 mmol) in dry DCM (2 ml) over 5 minutes, again maintaining the
temperature at below -60 C. The resulting white precipitous solution
was stirred for 30 mins at -78 C and treated, dropwise, with
triethylamine (3.43 ml, 2.49 g, 24.6 mmol, 10 eq.) before allowing the
resulting pale yellow suspension to come to room temperature. Water (20 ml)
was added, then the layers separated and the aqueous layer was extracted with
DCM (3 x 50 ml). The combined organic extracts were washed with saturated
NH4Cl (2 x 25ml) and brine (25 ml), dried (MgSO4), and the solvent removed
in vacuo to yield a yellow crystalline solid. The crude product was
purified by flash chromatography on silica gel (30 g in a 2 cm diameter
column) with ether as eluent to yield the diketone 24
(447 mg, 1.74 mmol, 71%) as colourless needles; m.p. 180 C
(ether/petrol); [
Preparation of
(2S,4R,5R,6R,8S,10R)
2,8-dimethyl-1,7,11,14-tetraoxa dispiro[4.0.4.4]tetradecan-4,10-diol 25.
To a solution of the diketone 24 (400 mg, 1.57 mmol) in THF (3
ml) was added lithium aluminium hydride (1.0 M in THF, 15.7 ml, 15.7 mmol, 10
eq.) dropwise at -78 C over 10 minutes. The resulting colourless
solution was allowed to come to room temperature and stirred for 18 hours. The
mixture was then recooled to -78 C and quenched by the addition of
sodium sulphate decahydrate (3 g). The mixture was allowed to come to room
temperature. Water (5 ml) and DCM (20 ml) were then added and the aqueous layer
was acidified with 3N hydrochloric acid (1ml). The layers separated and the
aqueous layer was extracted with DCM (3 x 50 ml). The combined organic
extracts were washed with saturated sodium bicarbonate (20 ml), dried (MgSO4),
and the solvent removed in vacuo to yield a yellow crystalline solid.
The crude product was purified by flash chromatography on silica gel (15 g in a
1 cm diameter column) with 75% ether in petrol as eluent to yield the diol
25 (359 mg, 1.38 mmol, 88%) as colourless blocks; m.p. 68 C
(ether/petrol); [
Preparation of (2R/S,4S)
2-Benzenesulfonyl-4-methyl-tetrahydrofuran 29.
To a solution of the lactone 28 (10.57 g, 106 mmol) in THF (500 ml) at
-78 C under argon was added DIBAL-H (1.5 M in toluene, 77.3 ml, 116
mmol, 1.1 eq.) dropwise over 15 minutes so as to maintain the temperature below
-60 C. The resulting colourless solution was stirred at -78 C
for 3 hours. The reaction was quenched by the addition of sodium sulfate
decahydrate (10 g) over 20 mins. The resulting suspension was allowed to come
to room temperature, stirred for 3 hours and filtered, washing with ethyl
acetate (300 ml). The filtrate was then concentrated in vacuo to
yield the crude lactol as a colourless oil.
To a solution of the crude lactol and calcium chloride (35.3 g, 318 mmol, 3
eq.) in dry DCM (300 ml) at room temperature under argon was added, in one
portion, benzene sulphinic acid (30.1 g, 212 mmol, 2 eq.). The resulting white
suspension was stirred at room temperature for 16 hours. The reaction was
filtered and the residues washed with DCM (400 ml). The filtrate was washed
with saturated sodium bicarbonate solution (2 x 150 ml), dried (MgSO4), and the
solvent removed in vacuo to yield a pale yellow oil. This crude product
was purified by flash column chromatography on silica gel (200 g in a 8 cm
diameter column) with 50% ether / petrol as eluent to yield the anomeric
sulfone 29 as an inseparable mixture of anomers in a 2.1:1 ratio
(15.3 g, 67.8 mmol, 64%) as a colourless oil;
Preparation of (3S,3'S)
3,3'-dimethyl-2,2',3,3'-tetrahydro-5,5'-bi-furan 5.
The anomeric sulphone 29 (5 g, 22.1 mmol) in THF (15 ml) was
added to a solution of magnesium bromide etherate (11.41 g, 44.2 mmol, 2 eq.)
and triethylamine (4.62 ml, 3.35 g, 33.1 mmol, 1.5 eq.) in THF (35 ml)
under argon. The heterogeneous mixture was sonicated for 2 hours after which
time no starting material remained by TLC. The flask was then fitted with a
distillation apparatus and all volatile components of the reaction mixture
distilled over. The distillate was then cooled to -78 C under argon
and treated with tert-butyllithium (1.7 M in pentanes, 12.1 ml, 20.7
mmol, 1.1 eq.). The resulting pale yellow solution was allowed to come to 0
C and stirred for 30 minutes. It was then cooled down to -78 C
and transferred by cannula to a solution of copper (II) chloride (2.78 g,
20.7 mmol, 1.1 eq) and palladium (II) chloride bisacetonitrile
complex (110 mg, 4.2 x 10-4 mol, 2.25 mol%) in THF (20 ml) at 0
C. The resulting black solution was stirred for 1 hour at 0
C and then quenched by the addition of 0.88 ammonia solution (6 ml) in
saturated ammonium chloride (24 ml) The resulting solution was stirred
vigorously at room temperature for 30 minutes to ensure the total oxidation of
ccopper (I) salts. The reaction mixture was then filtered through a pad of
Celitereg. washing with ether (20 ml). The aqueous layer was
extracted with ether (3 x 40 ml), the combined organic layers were dried
(MgSO4), and the solvent was removed in vacuo to yield a brown oil.
This crude product was purified by flash column chromatography on silica gel
(20 g in a 1 cm diameter column) with 2% ether in petrol as eluent to yield the
diene 5 (471 mg, 2.85 mmol, 26%) as a colourless oil; [
Preparation of
(3S,4S,5R,6R,9S,10S)
3,9-dimethyl-1,7,11,14-tetraoxadispiro[4.0.4.4]tetradecan-4,10-diol 30.
To an acetone solution of dimethyl dioxirane (c.a. 0.05 M, 250 ml) at
-78 C was added a solution of the bis-dihydrofuran 5 (300
mg, 1.80 mol) in ether (1 ml). The solution was stirred at this temperature
for 2 hours before being allowed to come to room temperature. The solvent was
then removed in vacuo and the resulting white solid suspended in toluene
(5 ml). Ethylene glycol (2 ml) and CSA (50 mg, cat.) were then added and the
solution heated at 120 C for 2 hours with vigorous stirring. The
resulting brown solution was allowed to come to room temperature and was poured
into saturated sodium bicarbonate solution (20 ml). The mixture was stirred
for 5 minutes and DCM (100 ml) was added. The organic layer was separated and
the aqueous layer extracted with DCM (3 x 100 ml). The combined organic phases
were dried (MgSO4), and the solvent removed in vacuo to furnish a yellow
oil. This crude product was purified by flash column chromatography on silica
gel (20 g in a 1 cm diameter column) with ether as eluent to yield the diol
30 (276 mg, 1.06 mmol, 59%) as colourless needles; m.p. 64 C
(ether/petrol); [
Experimental
 max
(film)/cm-1 2975, 1741 (C=O), 1190, 1125, 1063, 1011, 992;
max
(film)/cm-1 2975, 1741 (C=O), 1190, 1125, 1063, 1011, 992;  H
(200 MHz; CDCl3) (MAJOR ANOMER) 1.19 (3H, t, J 7.1,
CH3CH2O-), 1.37 (3H, d, J 6.3, 3 x H-3), 1.4-1.9 (6H, m, 2 x
H-3', 2 x H-4', 2 x H-5'), 3.42 (1H, m, H-2), 3.83 (2H, m, H-6'), 4.1 (2H,
q, J 7.0, CH3CH2O-), 4.6 (1H, m, H-2'); (MINOR ANOMER) 1.20 (3H,
t, J 7.0, CH3CH2O-), 1.32 (3H, d, J 6.5, 3 x H-3), 1.4-1.9
(6H, m, 2 x H-3', 2 x H-4'. 2 x H-5'), 3.40 (1H, m, H-2), 3.85 (2H, m, H-6'),
4.10 (2H, q, J 7.0, CH3CH2O-), 4.6 (1H, m, H-2'); C (50
MHz; CDCl3) (MAJOR ANOMER): 13.9 (CH3CH2O-), 18.4 (C-3), 18.9
(C-4'), 25.1 (C-5'), 30.1 (C-3'), 60.4 (C-2), 62.1 (C-6'), 69.7
(CH3CH2O-), 97.3 (C-2'), 173.0 (C-1); (MINOR ANOMER): 13.9
(CH3CH2O-), 17.7 (C-3), 18.8 (C-4'), 25.0 (C-5'), 30.1 (C-3'), 60.3
(C-2), 61.9 (C-6'), 72.2 (CH3CH2O-), 98.0 (C-2'), 172.9 (C-1);
m/z (EI) 202 (M)+, 184, 158, 144, 129, 101, 85, 67, 56, 45;
Found: (M)+, 202.1206. C10H18O4 requires: M, 202.1205.
H
(200 MHz; CDCl3) (MAJOR ANOMER) 1.19 (3H, t, J 7.1,
CH3CH2O-), 1.37 (3H, d, J 6.3, 3 x H-3), 1.4-1.9 (6H, m, 2 x
H-3', 2 x H-4', 2 x H-5'), 3.42 (1H, m, H-2), 3.83 (2H, m, H-6'), 4.1 (2H,
q, J 7.0, CH3CH2O-), 4.6 (1H, m, H-2'); (MINOR ANOMER) 1.20 (3H,
t, J 7.0, CH3CH2O-), 1.32 (3H, d, J 6.5, 3 x H-3), 1.4-1.9
(6H, m, 2 x H-3', 2 x H-4'. 2 x H-5'), 3.40 (1H, m, H-2), 3.85 (2H, m, H-6'),
4.10 (2H, q, J 7.0, CH3CH2O-), 4.6 (1H, m, H-2'); C (50
MHz; CDCl3) (MAJOR ANOMER): 13.9 (CH3CH2O-), 18.4 (C-3), 18.9
(C-4'), 25.1 (C-5'), 30.1 (C-3'), 60.4 (C-2), 62.1 (C-6'), 69.7
(CH3CH2O-), 97.3 (C-2'), 173.0 (C-1); (MINOR ANOMER): 13.9
(CH3CH2O-), 17.7 (C-3), 18.8 (C-4'), 25.0 (C-5'), 30.1 (C-3'), 60.3
(C-2), 61.9 (C-6'), 72.2 (CH3CH2O-), 98.0 (C-2'), 172.9 (C-1);
m/z (EI) 202 (M)+, 184, 158, 144, 129, 101, 85, 67, 56, 45;
Found: (M)+, 202.1206. C10H18O4 requires: M, 202.1205.
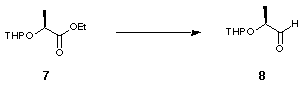 max (film)/cm-1
1952, 1733 (C=O), 1202, 1185, 1074, 903, 871; H (200 MHz; CDCl3)
(MAJOR ANOMER) 1.22 (3H, d, J 6.8, 3 x H-3), 1.4-1.9 (6H, m, 2 x
H-3', 2 x H-4' and 2 x H-5'), 3.7-3.8 (2H, m, 2 x H-6'), 3.93 (1H, dq, J
2.1, 6.9, H-2), 4.62 (1H, m, H-2'), 9.60 (1H, d, J 2.1, H-1);
(MINOR ANOMER) 1.31 (3H, d, J 7.1, 3 x H-3), 1.4-1.9 (6H, m, 2 x H-3', 2
x H-4' and 2 x H-5'), 3.7-3.8 (2H, m, 2 x H-6'), 4.19 (1H, dq, J 2.0,
7.0, H-2), 4.60 (1H, m, H-2'), 9.60 (1H, d, J 2.0, H-1); C (50
MHz; CDCl3) (MAJOR ANOMER): 14.2 (C-3), 18.9 (C-4'), 24.2 (C-5'), 29.8
(C-3'), 62.5 (C-6'), 75.6 (C-2), 98.0 (C-2'), 202.0 (C-1); (MINOR ANOMER): 14.7
(C-3), 18.4 (C-4'), 24.3 (C-5'), 30.0 (C-3'), 61.9 (C-6'), 77.6 (C-2), 98.7
(C-2'), 202.4 (C-1); m/z (EI) 157 (M-H)+, 143, 129, 101, 85,
56; Found: (M-H)+, 157.0867. C8H13O3 requires: M,
157.0865.
max (film)/cm-1
1952, 1733 (C=O), 1202, 1185, 1074, 903, 871; H (200 MHz; CDCl3)
(MAJOR ANOMER) 1.22 (3H, d, J 6.8, 3 x H-3), 1.4-1.9 (6H, m, 2 x
H-3', 2 x H-4' and 2 x H-5'), 3.7-3.8 (2H, m, 2 x H-6'), 3.93 (1H, dq, J
2.1, 6.9, H-2), 4.62 (1H, m, H-2'), 9.60 (1H, d, J 2.1, H-1);
(MINOR ANOMER) 1.31 (3H, d, J 7.1, 3 x H-3), 1.4-1.9 (6H, m, 2 x H-3', 2
x H-4' and 2 x H-5'), 3.7-3.8 (2H, m, 2 x H-6'), 4.19 (1H, dq, J 2.0,
7.0, H-2), 4.60 (1H, m, H-2'), 9.60 (1H, d, J 2.0, H-1); C (50
MHz; CDCl3) (MAJOR ANOMER): 14.2 (C-3), 18.9 (C-4'), 24.2 (C-5'), 29.8
(C-3'), 62.5 (C-6'), 75.6 (C-2), 98.0 (C-2'), 202.0 (C-1); (MINOR ANOMER): 14.7
(C-3), 18.4 (C-4'), 24.3 (C-5'), 30.0 (C-3'), 61.9 (C-6'), 77.6 (C-2), 98.7
(C-2'), 202.4 (C-1); m/z (EI) 157 (M-H)+, 143, 129, 101, 85,
56; Found: (M-H)+, 157.0867. C8H13O3 requires: M,
157.0865.
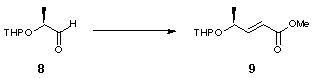 max (film)/cm-1
2948, 1731 (C=O), 1661, 1438, 1302, 1275, 1170, 1128, 1076, 1035; H (200
MHz; CDCl3) MAJOR ANOMER: 1.23 (3H, d, J 6.6, 3 x H-5), 1.3-1.9
(6H, m, 2 x H-3', 2 x H-4' and 2 x H-5'), 3.2-4.0 (3H, m, H-4 and 2 x H-6'),
3.65 (3H, s, MeO-), 4.42 (1H, m, H-2'), 5.85 (1H, dd, J 15.7, 1.4, H-2),
6.73 (1H, dd, J 15.7, 6.1, H-3); MINOR ANOMER: 1.16 (3H, d, J
6.6, 3 x H-5), 1.3-1.9 (6H, m, 2 x H-3', 2 x H-4' and 2 x H-5'), 3.2-4.0
(3H, m, H-4 and 2 x H-6'), 3.64 (3H, s, MeO-), 4.49 (1H, m, H-2'), 6.02 (1H, m,
H-2), 6.87 (1H, dd, J 15.7, 4.8, H-3); C (50 MHz; CDCl3)
MAJOR ANOMER: 18.9 (C-5), 21.0 (C-4'), 25.1 (C-5'), 30.4 (C-3'), 51.3
(OMe), 62.2 (C-6'), 70.4 (C-4), 96.6 (C-2'), 120.5 (C-2), 149.2 (C-3), 166.6
(C-1); MINOR ANOMER: 19.2 (C-5), 22.3 (C-4'), 25.2 (C-5'), 30.4 (C-3'), 51.3
(OMe), 62.0 (C-6'), 69.7 (C-4), 94.3 (C-2'), 118.7 (C-2), 150.0 (C-3), 166.9
(C-1); m/z (EI) 214 (M)+, 181, 169, 130, 113, 101, 85, 67,
55; Found: (M)+, 214.1192. C11H18O4 requires: M, 214.1205.
max (film)/cm-1
2948, 1731 (C=O), 1661, 1438, 1302, 1275, 1170, 1128, 1076, 1035; H (200
MHz; CDCl3) MAJOR ANOMER: 1.23 (3H, d, J 6.6, 3 x H-5), 1.3-1.9
(6H, m, 2 x H-3', 2 x H-4' and 2 x H-5'), 3.2-4.0 (3H, m, H-4 and 2 x H-6'),
3.65 (3H, s, MeO-), 4.42 (1H, m, H-2'), 5.85 (1H, dd, J 15.7, 1.4, H-2),
6.73 (1H, dd, J 15.7, 6.1, H-3); MINOR ANOMER: 1.16 (3H, d, J
6.6, 3 x H-5), 1.3-1.9 (6H, m, 2 x H-3', 2 x H-4' and 2 x H-5'), 3.2-4.0
(3H, m, H-4 and 2 x H-6'), 3.64 (3H, s, MeO-), 4.49 (1H, m, H-2'), 6.02 (1H, m,
H-2), 6.87 (1H, dd, J 15.7, 4.8, H-3); C (50 MHz; CDCl3)
MAJOR ANOMER: 18.9 (C-5), 21.0 (C-4'), 25.1 (C-5'), 30.4 (C-3'), 51.3
(OMe), 62.2 (C-6'), 70.4 (C-4), 96.6 (C-2'), 120.5 (C-2), 149.2 (C-3), 166.6
(C-1); MINOR ANOMER: 19.2 (C-5), 22.3 (C-4'), 25.2 (C-5'), 30.4 (C-3'), 51.3
(OMe), 62.0 (C-6'), 69.7 (C-4), 94.3 (C-2'), 118.7 (C-2), 150.0 (C-3), 166.9
(C-1); m/z (EI) 214 (M)+, 181, 169, 130, 113, 101, 85, 67,
55; Found: (M)+, 214.1192. C11H18O4 requires: M, 214.1205.
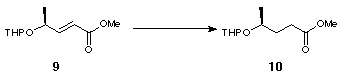 max (film)/cm-1
2942, 1738 (C=O), 1200, 1172, 1134, 1076, 1023, 997; H (200 MHz;
CDCl3) (MAJOR ANOMER) 1.14 (3H, d, J 6.8, 3 x H-5), 1.3-1.8 (8H,
m, 2 x H-3, 2 x H-3', 2 x H-4' and 2 x H-5'), 2.2-2.4 (2H, m, 2 x H-2), 3.3-3.5
(1H, m, H-4), 3.58 (3H, s, MeO-), 3.6-3.8 (2H, m, 2 x H-6'), 4.50 (1H, m,
H-2'); (MINOR ANOMER) 1.03 (3H, d, J 6.8, 3 x H-5), 1.3-1.8 (8H, m, 2 x
H-3, 2 x H-3', 2 x H-4' and 2 x H-5'), 2.2-2.4 (2H, m, 2 x H-2), 3.3-3.5 (1H,
m, H-4), 3.58 (3H, s, MeO), 3.6-3.8 (2H, m, 2 x H-6'), 4.58 (1H, m,
H-2'); C (50 MHz; CDCl3) (MAJOR ANOMER): 19.6 (C-5), 21.2
(C-4'), 25.2 (C-5'), 29.7 (C-3), 30.8 (C-3'), 31.2 (C-2), 51.3 (OMe), 62.4
(C-6'), 73.0 (C-4), 98.8 (C-2'), 173.8 (C-1); (MINOR ANOMER): 19.4 (C-5), 21.2
(C-4'), 25.2 (C-5'), 30.1 (C-3), 31.2 (C-3'), 32.1 (C-2), 51.3 (OMe), 62.1
(C-6'), 69.7 (C-4), 95.3 (C-2'), 173.9 (C-1); m/z (EI) 215
(M)+, 185, 159, 115, 101, 85, 67, 55, 45; Found: (M)+,
216.1352. C11H20O4 requires: M, 216.1361.
max (film)/cm-1
2942, 1738 (C=O), 1200, 1172, 1134, 1076, 1023, 997; H (200 MHz;
CDCl3) (MAJOR ANOMER) 1.14 (3H, d, J 6.8, 3 x H-5), 1.3-1.8 (8H,
m, 2 x H-3, 2 x H-3', 2 x H-4' and 2 x H-5'), 2.2-2.4 (2H, m, 2 x H-2), 3.3-3.5
(1H, m, H-4), 3.58 (3H, s, MeO-), 3.6-3.8 (2H, m, 2 x H-6'), 4.50 (1H, m,
H-2'); (MINOR ANOMER) 1.03 (3H, d, J 6.8, 3 x H-5), 1.3-1.8 (8H, m, 2 x
H-3, 2 x H-3', 2 x H-4' and 2 x H-5'), 2.2-2.4 (2H, m, 2 x H-2), 3.3-3.5 (1H,
m, H-4), 3.58 (3H, s, MeO), 3.6-3.8 (2H, m, 2 x H-6'), 4.58 (1H, m,
H-2'); C (50 MHz; CDCl3) (MAJOR ANOMER): 19.6 (C-5), 21.2
(C-4'), 25.2 (C-5'), 29.7 (C-3), 30.8 (C-3'), 31.2 (C-2), 51.3 (OMe), 62.4
(C-6'), 73.0 (C-4), 98.8 (C-2'), 173.8 (C-1); (MINOR ANOMER): 19.4 (C-5), 21.2
(C-4'), 25.2 (C-5'), 30.1 (C-3), 31.2 (C-3'), 32.1 (C-2), 51.3 (OMe), 62.1
(C-6'), 69.7 (C-4), 95.3 (C-2'), 173.9 (C-1); m/z (EI) 215
(M)+, 185, 159, 115, 101, 85, 67, 55, 45; Found: (M)+,
216.1352. C11H20O4 requires: M, 216.1361.
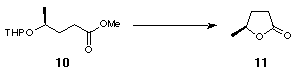 ]= -29.0 (c=
3.2, chloroform),[9] []= -29.6; max
(film)/cm-1 2942, 1738 (C=O), 1200, 1172, 1134, 1076, 1023, 997;
H (200 MHz; CDCl3) 1.33 (3H, d, J 7.1, Me-5), 1.72 (1H, m,
HAHB-4), 2.23 (1H, m, HAHB-4), 2.42 (2H, m, H-3), 4.58 (1H, m,
H-5); C (50 MHz; CDCl3) (20.8 (Me-5), 28.9 (C-4), 29.5 (C-3),
77.1 (C-5), 177.1 (C-2); m/z (EI) 100 (M)+, 85, 69, 56;
Found: (M)+, 100.0522. C5H8O2 requires: M, 100.0524.
]= -29.0 (c=
3.2, chloroform),[9] []= -29.6; max
(film)/cm-1 2942, 1738 (C=O), 1200, 1172, 1134, 1076, 1023, 997;
H (200 MHz; CDCl3) 1.33 (3H, d, J 7.1, Me-5), 1.72 (1H, m,
HAHB-4), 2.23 (1H, m, HAHB-4), 2.42 (2H, m, H-3), 4.58 (1H, m,
H-5); C (50 MHz; CDCl3) (20.8 (Me-5), 28.9 (C-4), 29.5 (C-3),
77.1 (C-5), 177.1 (C-2); m/z (EI) 100 (M)+, 85, 69, 56;
Found: (M)+, 100.0522. C5H8O2 requires: M, 100.0524.
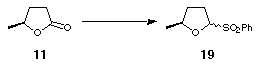 max (film)/cm-1 2975, 2932,
1584, 1446, 1384, 1309 (S=O), 1148 (S=O), 1074, 1044, 728, 689; H (200
MHz; CDCl3) (MAJOR ANOMER) 1.18 (3H, d, J 6.1, Me-5), 1.49 (1H,
m, HAHB-4), 2.1-2.6 (3H, 2 x H-3 and HAHB-4), 4.42 (1H, m, H-5),
4.90 (1H, dd, J 7.8, 4.9, H-2), 7.4-7.7 (3H, m, 2 x Hortho-Ar and
Hpara-Ar), 7.8-8.0 (2H, m, 2 x Hmeta-Ar); (MINOR ANOMER) 1.30
(3H, d, J 6.1, Me-5), 1.61 (1H, m, HAHB-4), 2.01 (1H, m,
HAHB-4), 2.1-2.6 (2H, m, 2 x H-3), 4.20 (1H, m, H-5), 4.80 (1H, dd, J
8.3, 2.3, H-2), 7.4-7.7 (3H, m, 2 x Hortho-Ar and Hpara-Ar),
7.8-8.0 (2H, m, 2 x Hmeta-Ar); m/z (EI) 226 (M)+, 161,
143, 125, 85 (M-PhSO2)+, 75, 67, 43, 29; Found: C, 58.50%; H, 6.35%;
S, 14.18%. C10H14O3S requires: C, 58.38%; H, 6.24%; S, 13.99%.
max (film)/cm-1 2975, 2932,
1584, 1446, 1384, 1309 (S=O), 1148 (S=O), 1074, 1044, 728, 689; H (200
MHz; CDCl3) (MAJOR ANOMER) 1.18 (3H, d, J 6.1, Me-5), 1.49 (1H,
m, HAHB-4), 2.1-2.6 (3H, 2 x H-3 and HAHB-4), 4.42 (1H, m, H-5),
4.90 (1H, dd, J 7.8, 4.9, H-2), 7.4-7.7 (3H, m, 2 x Hortho-Ar and
Hpara-Ar), 7.8-8.0 (2H, m, 2 x Hmeta-Ar); (MINOR ANOMER) 1.30
(3H, d, J 6.1, Me-5), 1.61 (1H, m, HAHB-4), 2.01 (1H, m,
HAHB-4), 2.1-2.6 (2H, m, 2 x H-3), 4.20 (1H, m, H-5), 4.80 (1H, dd, J
8.3, 2.3, H-2), 7.4-7.7 (3H, m, 2 x Hortho-Ar and Hpara-Ar),
7.8-8.0 (2H, m, 2 x Hmeta-Ar); m/z (EI) 226 (M)+, 161,
143, 125, 85 (M-PhSO2)+, 75, 67, 43, 29; Found: C, 58.50%; H, 6.35%;
S, 14.18%. C10H14O3S requires: C, 58.38%; H, 6.24%; S, 13.99%.
 ]=
-88 (c= 1.45, chloroform); max (film)/cm-1 2929, 2831, 1672,
1426, 1386, 1280, 1011, 790; H (200 MHz; CDCl3) 1.35 (6H, d, J
6.2, Me-2 and Me-2'), 2.31 (2H, ddd, J 15.0, 8.0, 2.0, HAHB-3
and HAHB-3'), 2.82 (2H, ddd, J 14.7, 11.7, 1.6, HAHB-3 and
HAHB-3'), 4.72 (2H, m, H-2 and H-2'), 5.03 (2H, t, J 2.2, H-4 and
H-4'); C (50 MHz; CDCl3) 21.7 (Me-2 and Me-2'), 37.5 (C-3 and C-3'),
78.2 (C-2 and C-2'), 97.4 (C-4 and C-4'), 148.0 (C-5 and C-5'); m/z (EI)
166 (M)+, 138, 111, 83, 55; Found: (M)+ 166.0993.
C10H14O2 requires: M, 166.0994.
]=
-88 (c= 1.45, chloroform); max (film)/cm-1 2929, 2831, 1672,
1426, 1386, 1280, 1011, 790; H (200 MHz; CDCl3) 1.35 (6H, d, J
6.2, Me-2 and Me-2'), 2.31 (2H, ddd, J 15.0, 8.0, 2.0, HAHB-3
and HAHB-3'), 2.82 (2H, ddd, J 14.7, 11.7, 1.6, HAHB-3 and
HAHB-3'), 4.72 (2H, m, H-2 and H-2'), 5.03 (2H, t, J 2.2, H-4 and
H-4'); C (50 MHz; CDCl3) 21.7 (Me-2 and Me-2'), 37.5 (C-3 and C-3'),
78.2 (C-2 and C-2'), 97.4 (C-4 and C-4'), 148.0 (C-5 and C-5'); m/z (EI)
166 (M)+, 138, 111, 83, 55; Found: (M)+ 166.0993.
C10H14O2 requires: M, 166.0994.
 ]= -112 (c= 0.38, chloroform); max (film)/cm-1 3526
(OH), 2968, 2927, 1384, 1078, 966, 917; H (200 MHz; CDCl3) 1.22
(6H, d, J 6.3, Me-2 and Me-8), 1.81 (2H, ddd, J 12.6, 8.5, 5.6,
HAHB-3 and HAHB-9), 2.08 (2H, m, HAHB-3 and
HAHB-9), 2.66 (2H, d, J 4.0, 2 x OH), 3.60 (2H, m, Heq-12
and Heq-13), 4.08 (2H, ddd, J 12.2, 8.5, 3.9, H-4 and H-10), 4.20 (2H,
m, Hax-12 and Hax-13), 4.32 (2H, m, H-2 and H-8); C (50 MHz; CDCl3)
21.5 (Me-2 and Me-8), 38.7 (C-3 and C-9), 59.9 (C-12 and C-13), 72.3, 72.8
(C-2 and C-8 and C-4 and C-10), 101.4 (C-5 and C-6); m/z (EI) 260
(M)+, 245 (M-CH3)+, 216, 200 (M-C2H4O2)+, 189,
144, 118, 99 (C6H11O)+, 42, 57; Found: C, 55.41%; H, 7.82%.
C12H20O6 requires: C, 55.36%; H, 7.75%.
]= -112 (c= 0.38, chloroform); max (film)/cm-1 3526
(OH), 2968, 2927, 1384, 1078, 966, 917; H (200 MHz; CDCl3) 1.22
(6H, d, J 6.3, Me-2 and Me-8), 1.81 (2H, ddd, J 12.6, 8.5, 5.6,
HAHB-3 and HAHB-9), 2.08 (2H, m, HAHB-3 and
HAHB-9), 2.66 (2H, d, J 4.0, 2 x OH), 3.60 (2H, m, Heq-12
and Heq-13), 4.08 (2H, ddd, J 12.2, 8.5, 3.9, H-4 and H-10), 4.20 (2H,
m, Hax-12 and Hax-13), 4.32 (2H, m, H-2 and H-8); C (50 MHz; CDCl3)
21.5 (Me-2 and Me-8), 38.7 (C-3 and C-9), 59.9 (C-12 and C-13), 72.3, 72.8
(C-2 and C-8 and C-4 and C-10), 101.4 (C-5 and C-6); m/z (EI) 260
(M)+, 245 (M-CH3)+, 216, 200 (M-C2H4O2)+, 189,
144, 118, 99 (C6H11O)+, 42, 57; Found: C, 55.41%; H, 7.82%.
C12H20O6 requires: C, 55.36%; H, 7.75%.
 ]= -151 (c= 1.48, chloroform); max
(film)/cm-1 2975, 2930, 1758 (C=O), 1104, 1088, 1061; H (200
MHz; CDCl3) 1.38 (6H, d, J 5.1, Me-2 and Me-8), 2.15 (2H, dd,
J 18.0, 9.4, HAHB-3 and HAHB-9), 2.72 (2H, dd, J 18.0,
5.7, HAHB-3 and HAHB-9), 3.89 (2H, m, Heq-12 and Heq-13), 4.25
(2H, m, Hax-12 and Hax-13), 4.58 (2H, m, H-2 and H-8); C (50 MHz;
CDCl3) 21.1 (Me-2 and Me-8), 43.6 (C-3 and C-9), 62.0 (C-12 and C-13),
71.6 (C-2 and C-8), 95.0 (C-5 and C-6), 206.1 (C-4 and C-10); m/z (EI)
256 (M)+, 187, 142, 115, 83, 73, 49; Found: (M)+,
256.0954. C12H16O6 requires: M, 256.0947.
]= -151 (c= 1.48, chloroform); max
(film)/cm-1 2975, 2930, 1758 (C=O), 1104, 1088, 1061; H (200
MHz; CDCl3) 1.38 (6H, d, J 5.1, Me-2 and Me-8), 2.15 (2H, dd,
J 18.0, 9.4, HAHB-3 and HAHB-9), 2.72 (2H, dd, J 18.0,
5.7, HAHB-3 and HAHB-9), 3.89 (2H, m, Heq-12 and Heq-13), 4.25
(2H, m, Hax-12 and Hax-13), 4.58 (2H, m, H-2 and H-8); C (50 MHz;
CDCl3) 21.1 (Me-2 and Me-8), 43.6 (C-3 and C-9), 62.0 (C-12 and C-13),
71.6 (C-2 and C-8), 95.0 (C-5 and C-6), 206.1 (C-4 and C-10); m/z (EI)
256 (M)+, 187, 142, 115, 83, 73, 49; Found: (M)+,
256.0954. C12H16O6 requires: M, 256.0947.
 ]= -77.3 (c= 1.0, chloroform); max
(film)/cm-1 3365 (OH), 2968, 2927, 1132, 1080, 960; H (200
MHz; CDCl3) 1.40 (6H, d, J 6.2, Me-2 and Me-8), 1.58 (2H, m,
HAHB-3 and HAHB-9), 2.53 (2H, dt, J 13.9, 6.9,
HAHB-3 and HAHB-9), 3.49 (2H, m, Heq-12 and Heq-13), 3.98 (2H,
bs, 2 x OH), 4.12 (2H, m, Hax-12 and Hax-13), 4.20 (4H, m, H-2, H-4, H-8
and H-10); C (50 MHz; CDCl3) 21.1 (Me-2 and Me-8), 41.3 (C-3 and
C-9), 59.5 (C-12 and C-13), 73.4 (C-2 and C-8), 79.6 (C-4 and C-10), 105.3 (C-5
and C-6); m/z (EI) 260 (M)+, 245 (M-CH3)+, 216,
200 (M-C2H4O2)+, 189, 144, 118, 99 (C6H11O)+, 42, 57;
Found: C, 55.61%; H, 7.89%. C12H20O6 requires: C, 55.36%; H, 7.75%.
]= -77.3 (c= 1.0, chloroform); max
(film)/cm-1 3365 (OH), 2968, 2927, 1132, 1080, 960; H (200
MHz; CDCl3) 1.40 (6H, d, J 6.2, Me-2 and Me-8), 1.58 (2H, m,
HAHB-3 and HAHB-9), 2.53 (2H, dt, J 13.9, 6.9,
HAHB-3 and HAHB-9), 3.49 (2H, m, Heq-12 and Heq-13), 3.98 (2H,
bs, 2 x OH), 4.12 (2H, m, Hax-12 and Hax-13), 4.20 (4H, m, H-2, H-4, H-8
and H-10); C (50 MHz; CDCl3) 21.1 (Me-2 and Me-8), 41.3 (C-3 and
C-9), 59.5 (C-12 and C-13), 73.4 (C-2 and C-8), 79.6 (C-4 and C-10), 105.3 (C-5
and C-6); m/z (EI) 260 (M)+, 245 (M-CH3)+, 216,
200 (M-C2H4O2)+, 189, 144, 118, 99 (C6H11O)+, 42, 57;
Found: C, 55.61%; H, 7.89%. C12H20O6 requires: C, 55.36%; H, 7.75%.
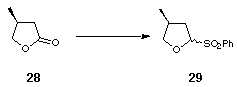 max (film)/cm-1
2983, 2955, 1587, 1444, 1385, 1306 (S=O), 1149 (S=O), 1076, 1044, 741, 687;
H (200 MHz; CDCl3) (MAJOR ANOMER) 1.04 (3H, d, J 6.3,
Me-4), 1.8-3.0 (3H, m, 2 x H-3 and H-4), 3.52 (1H, dd, J 17.9, 12.1,
HAHB-5), 4.22 (1H, dd, J 12.1, 9.2, HAHB-5), 4.92 (1H,
dd, J 8.1, 3.9, H-2), 7.5-7.7 (3H, m, 2 x Hortho-Ar and
Hpara-Ar), 7.9-8.0 (2H, m, 2 x Hmeta-Ar); (MINOR ANOMER) 1.12
(3H, d, J 6.1, Me-4), 2.1-2.9 (3H, m, 2 x H-3 and H-4), 3.52 (1H, dd,
J 11.8, 2.1, HAHB-5), 4.03 (1H, dd, J 11.7, 8.3,
HAHB-5), 4.92 (1H, dd, J 7.6, 3.8, H-2), 7.5-7.7 (3H, m, 2 x
Hortho-Ar and Hpara-Ar), 7.9-8.0 (2H, m, 2 x Hmeta-Ar);
C (50 MHz; CDCl3) (MAJOR ANOMER) 17.7 (Me-4), 32.7 (C-4), 33.9
(C-3), 76.3 (C-5), 94.0 (C-2), 128.9 129.2 (2 x Cmeta-Ar and 2 x
Cortho-Ar), 133.8 (Cpara-Ar), 137.1 (Cipso-Ar); (MINOR
ANOMER) 15.0 (Me-4), 33.1 (C-4), 33.9 (C-3), 77.2 (C-5), 94.3 (C-2), 128.9
129.2 (2 x Cmeta-Ar and 2 x Cortho-Ar), 133.8 (Cpara-Ar),
136.9 (Cipso-Ar); m/z (EI) 226 (M)+, 161, 143, 125, 85
(M-PhSO2)+, 75, 67, 43, 29; Found: (M)+ 226.0661.
C10H14O3S requires: M, 226.0664.
max (film)/cm-1
2983, 2955, 1587, 1444, 1385, 1306 (S=O), 1149 (S=O), 1076, 1044, 741, 687;
H (200 MHz; CDCl3) (MAJOR ANOMER) 1.04 (3H, d, J 6.3,
Me-4), 1.8-3.0 (3H, m, 2 x H-3 and H-4), 3.52 (1H, dd, J 17.9, 12.1,
HAHB-5), 4.22 (1H, dd, J 12.1, 9.2, HAHB-5), 4.92 (1H,
dd, J 8.1, 3.9, H-2), 7.5-7.7 (3H, m, 2 x Hortho-Ar and
Hpara-Ar), 7.9-8.0 (2H, m, 2 x Hmeta-Ar); (MINOR ANOMER) 1.12
(3H, d, J 6.1, Me-4), 2.1-2.9 (3H, m, 2 x H-3 and H-4), 3.52 (1H, dd,
J 11.8, 2.1, HAHB-5), 4.03 (1H, dd, J 11.7, 8.3,
HAHB-5), 4.92 (1H, dd, J 7.6, 3.8, H-2), 7.5-7.7 (3H, m, 2 x
Hortho-Ar and Hpara-Ar), 7.9-8.0 (2H, m, 2 x Hmeta-Ar);
C (50 MHz; CDCl3) (MAJOR ANOMER) 17.7 (Me-4), 32.7 (C-4), 33.9
(C-3), 76.3 (C-5), 94.0 (C-2), 128.9 129.2 (2 x Cmeta-Ar and 2 x
Cortho-Ar), 133.8 (Cpara-Ar), 137.1 (Cipso-Ar); (MINOR
ANOMER) 15.0 (Me-4), 33.1 (C-4), 33.9 (C-3), 77.2 (C-5), 94.3 (C-2), 128.9
129.2 (2 x Cmeta-Ar and 2 x Cortho-Ar), 133.8 (Cpara-Ar),
136.9 (Cipso-Ar); m/z (EI) 226 (M)+, 161, 143, 125, 85
(M-PhSO2)+, 75, 67, 43, 29; Found: (M)+ 226.0661.
C10H14O3S requires: M, 226.0664.
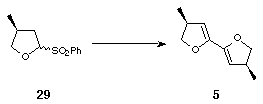 ]=
-75 (c=0.56, chloroform); max (film)/cm-1 2975, 2930, 2875,
1203, 1079, 913, 905, 738, 790; H (200 MHz; CDCl3) 1.09 (6H,
d, J 6.8, Me-3, Me-3'), 3.13 (2H, m, H-3 and H-3'), 3.95 (2H, dd, J
8.6, 6.7, HAHB-2 and HAHB-2'), 4.46 (2H, dd, J 9.5,
8.7, HAHB-2 and HAHB-2'), 5.14 (2H, d, J 2.3, H-4 and
H-4'); C (50 MHz; CDCl3) 20.0 (Me-3 and Me-3'), 37.6 (C-3 and C-3'),
77.2 (C-2 and C-2'), 104.9 (C-4 and C-4'), 148.0 (C-5 and C-5'); m/z
(EI) 166 (M)+, 153, 129, 113, 97, 86, 84, 59, 49, 47; Found:
(M)+ 166.1005. C10H14O2 requires: M, 166.0994.
]=
-75 (c=0.56, chloroform); max (film)/cm-1 2975, 2930, 2875,
1203, 1079, 913, 905, 738, 790; H (200 MHz; CDCl3) 1.09 (6H,
d, J 6.8, Me-3, Me-3'), 3.13 (2H, m, H-3 and H-3'), 3.95 (2H, dd, J
8.6, 6.7, HAHB-2 and HAHB-2'), 4.46 (2H, dd, J 9.5,
8.7, HAHB-2 and HAHB-2'), 5.14 (2H, d, J 2.3, H-4 and
H-4'); C (50 MHz; CDCl3) 20.0 (Me-3 and Me-3'), 37.6 (C-3 and C-3'),
77.2 (C-2 and C-2'), 104.9 (C-4 and C-4'), 148.0 (C-5 and C-5'); m/z
(EI) 166 (M)+, 153, 129, 113, 97, 86, 84, 59, 49, 47; Found:
(M)+ 166.1005. C10H14O2 requires: M, 166.0994.
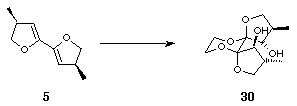 ]= -134 (c= 1.1, chloroform); max
(film)/cm-1 3400-3200 (OH), 2929, 2831, 1672, 1426, 1386, 1280,
1011, 790; H (200 MHz; CDCl3) 1.15 (6H, d, J 6.8, Me-3 and
Me-9), 2.38 (2H, m, H-3 and H-9), 2.55 (2H, bs, OH), 3.49 (2H, t, J
8.8, HAHB-2 and HAHB-8), 3.65 (2H, m, HAHB-2 and
HAHB-8), 3.65 (2H, m, Heq-12 and Heq-13), 4.14 (2H, m, H-4 and H-10),
4.25 (2H, m, Hax-12 and Hax-13); C (50 MHz; CDCl3) 15.3 (Me-3 and Me-9),
37.7 (C-3 and C-9), 60.0 (C-12 and C-13), 72.0 (C-2 and C-8), 78.6 (C-4 and
C-10), 101.3 (C-5 and C-6); m/z (EI) 260 (M)+, 245
(M-CH3)+, 216, 200 (M-C2H4O2)+, 189, 144, 118, 99
(C6H11O)+, 42, 57; Found: C, 55.37%; H, 7.79%. C12H20O6 requires:
C, 55.36%; H, 7.75%.
]= -134 (c= 1.1, chloroform); max
(film)/cm-1 3400-3200 (OH), 2929, 2831, 1672, 1426, 1386, 1280,
1011, 790; H (200 MHz; CDCl3) 1.15 (6H, d, J 6.8, Me-3 and
Me-9), 2.38 (2H, m, H-3 and H-9), 2.55 (2H, bs, OH), 3.49 (2H, t, J
8.8, HAHB-2 and HAHB-8), 3.65 (2H, m, HAHB-2 and
HAHB-8), 3.65 (2H, m, Heq-12 and Heq-13), 4.14 (2H, m, H-4 and H-10),
4.25 (2H, m, Hax-12 and Hax-13); C (50 MHz; CDCl3) 15.3 (Me-3 and Me-9),
37.7 (C-3 and C-9), 60.0 (C-12 and C-13), 72.0 (C-2 and C-8), 78.6 (C-4 and
C-10), 101.3 (C-5 and C-6); m/z (EI) 260 (M)+, 245
(M-CH3)+, 216, 200 (M-C2H4O2)+, 189, 144, 118, 99
(C6H11O)+, 42, 57; Found: C, 55.37%; H, 7.79%. C12H20O6 requires:
C, 55.36%; H, 7.75%.