

 Copper Reagents: Do Higher Order Cuprates Exist?
Copper Reagents: Do Higher Order Cuprates Exist?
Cyanide-containing lithium cuprates (e.g. Me2CuLi*LiX; X = CN) complement
Gilman and homocuprates as important reagents in synthetic chemistry for
forming carbon-carbon bonds.1 Over the past decade, one school of
thought has claimed the chemically active species to contain tricoordinate,
dianionic Cu(I) in which cyanide is sigma-bonded to the metal
(i.e.[R2Cu-CN]-2, 2Li+, 1 or 2, latter solvated at Li by Me2O).
2,3,4
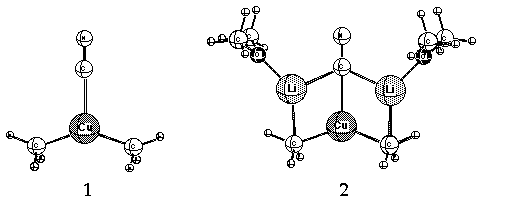
An amendment to the hypothesis suggested pi-bonding instead.5
Another viewpoint, based on NMR,5, 6 EXAFS spectroscopy
8.9
and ab initio calculations, 9 -11 asserts the non-existence of
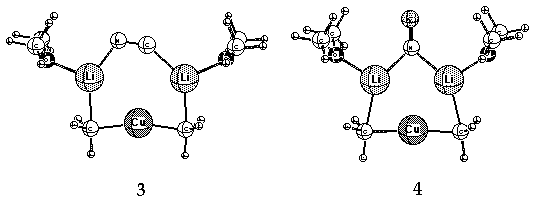
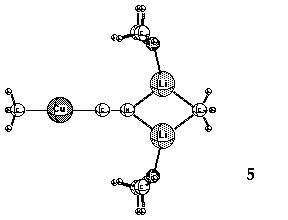
a Cu-CN bond in these complexes. Small rings (3, 4 and 5, Li cations solvated by Me2O)
containing lower-order, dicoordinate Cu(I) have been suggested as alternative,
more stable structures. 11
On the reactivity issue, there are two intruiging questions that remain open.
First, although various arguments for superior reactivity of HO cyanocuprates
have been made,1 a recent head-to-head study of conjugate addition
with cuprates prepared from CuCN and CuI indicates very little difference in
product yield.12 Second, whatever the structure of the Me2CuLi*LiCN
constitution, no convincing evidence for such an aggregate on the pathway to
the rate-determining transition state has yet been reported.
What is the structure of R2CuLi*LiCN? Is it possible to assign a
meaningful mechanistic role to any of the suggested structures in the chemistry
of the cuprate reagents? Can special reactivity be attributed to the presence
of cyanide vs halide (X = Hal)? Might the answer to these questions lead to
the design of other useful cuprate reagents?
Comments were added using this form during the conference
sessions.
Henry Rzepa, accepted 21/Jun/1996
Being unfamiliar with the literature on this subject, I
wonder if anyone can enlighten me on whether unsual
structural techniques such as STM (scanning Tunnelling
Microscopy) are applicable or have been used for this
structural problem?
Brian James, accepted 09/Jul/1996
There will probably never be a definitive solution structure known.
The fact is that there is no direct way of determining the solution structure.
The sprectral data is open to interpretation. The calculations are probably
the best data that we have. Mechanisms for Gilman Cuprates have been proposed
based on kinetic data. The question we need address about 'HO' cuprates then
is: Are they fudamentally different from Gilman Cuprates and if so How?
Steve Bertz, accepted 20/Jul/1996
Three techniques (NMR, EXAFS, theoretical calculations) have been brought to bear on the
higher order cyanocuprate problem, and they seem to be converging on the
same conclusion: higher order cyanocuprates do not exist.
(There is one higher order cuprate, Ph3CuLi3+, first prepared by
Bertz and Dabbagh and confirmed by Power and Olmstead using X-ray crystallography.)
No definitive characterization of either sigma- or pi-bound CN has been published.
The spectroscopic data on the 3pro2 side is fragmentary and inconclusive.
The conclusions that have been drawn from this data are non sequitors.
Professor Penner-Hahn, who did the EXAFS studies in collaboration with Professor Knochel,
informs me that he has done an IR study, but I have not received a preprint.
Would it be possible to get a precis here?
Professor van Koten tells me that he has also done EXAFS work, but
I have not received any details. Would it be possible for Gerard to
disclose his conclusions here? Before closing, it should be mentioned
that the calculations were done by Dr. J.P. Snyder and Professor G. Frenking.
Have any new theoretical insights appeared recently?
Jim Snyder, accepted 22/Jul/1996
Henry Rzepa has asked if STM or similar techniques have been applied
to the HO problem. Just to remind, no cyanide-containing aggregate with HO
structure has yet been isolated for X-ray study or solid surface evaluation.
We are presently stuck with species in ether-like solvents at -80 deg
or so whose existence has been inferred from nmr and ir spectra. As
mentioned by Steve Bertz, EXAFS has been applied by Penner-Hahn's group. This
solution-based technique suggests the absence of a third ligand to copper and
thus appears to rule out the tricoordinate [R2Cu-CN] moiety.
As to Brian James' question on the difference between lower-order Gilman
cuprates and the putative HO species, the low-energy results of the
calculations (structures 3, 4 and 5 above) suggest an intimate relationship.
That is, the best candidates (3 and 4) involve a Gilman cuprate [CH3-Cu-CH3]-
bridged formally by Li2CN+. This is equivalent to the latter being bridged by
[CH3-CU-CH3]Li2+, i.e. the now classic Gilman dimer. While the distribution of ions
in the last two sentences is a formal one - only to show the relationship - there is
an important geometric distinction. The Gilman dimers (aliphatic
and aromatic) are non-planar; the cyanide bridged species 3 and 4 are planar.
If the latter aggregates are on the reaction pathway to chemical products,
this spatical difference may contribute to reactivity. Likewise, if there are
subsequent specie on the reaction path (e.g. a tetracoordinate square planar copper
species containing CN; JACS 1995, 117, 11025), aggregates 3 and 4 might just
deliver the CN without a serious expenditure of entropy. At the moment, these
notions on reactivity are simply speculation. The energetic differences between
HO and bridged species derived from the calculations, however, are large and
unambiguous. This permits the reasonable conclusion that the initial composition
following reagent mixture is bridged-Gilman and not HO, whatever the course of
the subsequent reaction path. The ir, nmr and EXAFS measurements
- ground state techniques - are supportive.
Jim Snyder, accepted 23/Jul/1996
CORRECTION. In the middle of the last paragraph, I meant 'former' and
not 'latter'. That is, the formal ion pair [Me-Cu-Me]-[Li2CN]+ is
equivalent to [Me-Cu-Me]-[Li2(Me-Cu-Me)]+. One anion is simply being
replaced by another; i.e. CN- for [Me-Cu-Me]- with the geometric
consequences mentioned. Obviously, other anions (e.g. SCN-,
halides, etc) can in principle operate similarly. Equally obviously,
the actual charge in such an aggregate is much more evenly distributed
both in space and among the subset of ions.
Bruce H. Lipshutz, accepted 23/Jul/1996
The question as to the structure of higher order cuprates seems
to have generated considerable interest. What appears to be the case
is that whatever their structure, they are unique relative to prior
art. Clearly, with so many talented groups (Power, Boche, van Koten,
etc. having tried to obtain X-ray data without success, there must be
something about these reagents thatis, at least, 'different'. Those
who have presented in the literature experimental data claiming that
these species are dicoordinate, have made a reasonably comelling case.
And the calculations done by Snyder have served to bolster these
arguments. We, of course, do very much appreciate this data, for our
goal has always been to understand these reagents which we introduced
years ago. Should definitive experiments establish the position of the
cyanide ligand, and hence a likely structure result, we would be the
first to offer our congratulations. But at this point in time, it is
not clear to me, calculations notwithstanding, that the experimental
dat is foolproof. I am not swayed by NMR work, as coupling constants,
or lack thereof, are just not the answer. EXAFS and XANES can be
misleading, as commented to me some time ago by Hodgson at Stanford,
and Galen Stucky here at UCSB. On the other hand, we would take stock
in data obtained from IR experiments, which must reveal the presence,
or lack thereof, of a copper-carbon stretch. Most crucial in this
regard is the stretch associated with the Cu-C of the copper-cyanide
carbon. We have now purchased an IR instrument that will allow us to
establish, on the timescale of a molecular vibration, if this bond
remains intact in going from the 1:1 RLi:CuCN ratio, to the 2:1 ratio.
To our way of thinking, this appears to be the most indicative
experiment which we can devise, short of an X-ray, to address the
higher order question. To further complicate matters, however, is the
solvent effect, since it is likely that the species in THF is
different from that in Et2O, as we pointed out years ago. Hence, it is
possible that differences will materialize in these IR studies as a
function of medium. Lastly, I can offer a preliminary comment
regarding some experiments that we have done with nRLi + CuCN, looking
at their Raman spectra. Although done only twice on the MeLi
combinations, we see the same band in both the 1:1 and 2:1 ratios (at
440 cm-1). I do not wish to make too much of this right now, since we
are still learning how to use the equipment located in the Engineering
Department, and have not looked at other species, etc. But the band
seems to be in about the right place, as I understand it, but I could
use some comments on this technique for sure (it is our first effort
on this instrument).
Jim Snyder, accepted 24/Jul/1996
While this discussion is centered primarily around
cyanide-containing lithium cuprates (cf JOC 1994, 59, 2665 for a
definition), an ironic footnote to the decade-long debate is Steve
Bertz's standing as the originator of what ha s been interpreted as
the single structurally characterized 'HO' cuprate: Ph3CuLi3+ (JACS
1988, 110, 3668). Two variations on this compound have successfu lly
yielded to X-ray crystallography (Olmstead and Power JACS 1990, 112,
8008). The structures
portray Cu(I) embedded in a matrix of 3 phenyl anions (ergo, HO) and 3
lithium c ations. Analysis of this structure by ab initio calculations
suggests that the Cu(I) doe s not qualify as higher-order. A more
satisfying description views the structures as rare examples of the
phenyl lithium trimer templated electrostatically around the rather
isolated Cu(I) cation. One consequence of this interpretation is that
the central ion should be exchangeable by other cations of similar
size. Preliminary nmr experiments we have carried out suggest this to
be the case.
Jim Penner-Hahn, accepted 24/Jul/1996
We agree with Brian James that determination of solute structures is much
more difficult than determination of solid state structures and will thus
always be less definitive than crystallography. Nevertheless, there are
a variety of spectroscopic methods which can provide pieces of the
picture; Taken together, these can provide an informative view of the
solute structure.
In the case of the cyanocuprates, three spectroscopic probes have been
particularly informative: NMR, EXAFS, and IR. As summarized by Jim
Snyder in his initial commentary, interpretation of the NMR data has been
controversial. These data have been interpreted as providing evidence
for 'higher-order' cuprates and as providing evidence against
'higher-order' cuprates. In our view, this controversy is due to the
difficulty of assigning with confidence the structural interpretation of
a particular NMR chemical shift. All of the NMR studies of cyanocuprates
appear to agree that 1) There is a single dominant species formed from
CuCN+2RLi; and 2) The cyanide ion in this species is not identical to LiCN.
As mentioned by Steve Bertz, we have used extended x-ray absorption fine
structure (EXAFS) spectroscopy to characterize the Cu environment in
cyanocuprate solutions. EXAFS is in many regards a very limited
technique. It only provides information about atoms within about 5
angstroms of the absorber (Cu in this case) and it provides only limited
angular information. Perhaps most importantly, EXAFS can only provide
average structural information. However, with the NMR evidence for a
single dominant structure in solution, the averaging problem becomes less
severe. The attraction of EXAFS is that it is uniquely able to provide
bond lengths and coordination numbers with near crystallographic
precision. The EXAFS results demonstrate that the Cu in CuCN+2RLi is two
coordinate and that cyanide is not coordinated to the Cu (at least not in
a linear Cu-C-N structure).
The remaining spectroscopic evidence that bears on cuprate structure has
been IR spectroscopy. There are several reports [Lipshutz, et al, J. Org.
Chem. (1984) 49, 3943; J. Am. Chem. Soc. (1990) 112, 4032; Singer, & Oehlschla
ger,
J. Org. Chem. (1992) 57, 2192-2195] that the CN stretch occurs at the same frequ
ency in
both 'higher-order' and 'lower-order' cuprates. This is inconsistent with
our proposal that the cyanide is displaced from Cu on addition of the
second equivalent of RLi [In fact, this observation is probably also
inconsistent with the proposed trigonal Cu in 'higher-order' cuprates,
since the CN stretching frequency typically decreases by 30 cm-1 on
going from 2 to 3-coordinate Cu]. To address this question, we have
re-examined the IR spectra of the CuCN.2LiCl + nMeLi (n=0,3) using
low-temperature, high-resolution FTIR (Huang et al, J. Am. Chem. Soc.,
accepted for publication). We find that, in contrast with earlier
reports, CuCN.2LiCl + MeLi and CuCN.2LiCl + 2MeLi have distinct cyanide
stretches at 2133 cm-1 and 2115 cm-1, respectively. The earlier reports
(of only the 2133 cm-1 stretch) appear to be do to the fact that there is
an equilibrium 'LiMeCuCN' + MeLi ='Me2Cu.Li2CN' such that both species are
present when 2 equivalents of MeLi have been added.
The former only disappears when 2.2 equivalents of MeLi have been added.
Complicating this issue is the fact
that the oscillator strength for the CN stretch is significantly lower in
'Me2Cu.Li2CN' than in 'LiMeCuCN' or CuCN itself. The combination of an
equilibrium and an intrinsically weak transition make it possible to miss
the presence of a new CN stretch in the 2:1 solution.
Jim Snyder has calculated the expected frequencies and intensities for
the cyanide ion in different environments and finds excellent agreement
with structures such as 3 (these are described in the above cited IR
paper). In particular, the very weak CN stretch in 'Me2Cu.Li2CN' is
diagnostic of a cyanide that is bound in an ionic, non-polarizable
environment. This is completely consistent with a cyanide that interacts
with Li cations, but is inconsistent with a Cu-CN adduct.
In conclusion, each of the three spectroscopic techniques provides only a
piece of the picture of the overall structure. However, the three taken
together, particularly when combined with theoretical calculations, provide a
substantially complete picture of the structure of the CuCN.2LiCl+2RLi
reagent. In our view, this structure is most consistent with the
structure shown as 3 in the initial commentary.
Brian James, accepted 25/Jul/1996
There is a vast amount of IR and NMR data available which attemp
to address the structure of the 'HO' cuprate. EXAFS, XANES, and
Computational data have recently been added to the rapidly growing
body of data surrounding the existence/nonexistence of the HO cuprate.
Electrospray Mass Spectrometry has recently been applied to the
problem as well. Given this vast amount of data, why hasn't a single
hypothesis emerged. The reason is simple. All of the data is open to
interpretation, which is personally biased. I have been spending a
great deal of time mulling over the ideas that all of the other
contributors have published. This topic is going to make up a good
size portion of my future dissertation. I look at all of the data and
try to remain objective. All of the arguments are good, but maybe we
are all wrong and the answer is something that we haven't even come
close to, or maybe we are all right and the 'truth' is a
conglomeration of all of the ideas. Perhaps the thing that has been
bothering me the most lately is that everybody is arguing, for or
against a single structure. We need to address the fact that cuprates
do not exist as monomers in solution. We need to address the
aggregation of the species which are present. ESIMS has shown that
cuprate species exist in a vaiety of aggregation states, but there is
no way that I am aware of to quantify the data so as to state the
relative amounts of species present. Similarly there is no way to even
detect the neutral species that are most likely present. The
aggregation phenomenon is relevant to the arguments that everybody is
making, but nobody seems to be addressing it, why? How can we even
attempt to address it? What effect does it have on reactivity, when
there are 1, 2, or 10+ species in the flask when we add the substrate?
Jim Snyder, accepted 25/Jul/1996
It is delightful and gratifying that in this knotty area of cuprate structure and
reactivity, a graduate student - Brian James - is asking core questions. As teachers and
experienced researchers, the rest of us are 'supposed' to have the answers. In the following, I fall
into the teacher-trap of offering more answers than questions ...
Brian asks why no single hypothesis on the HO question has emerged. It
seems important to clarify what might fall under the umbrella of the all-embracing term 'hypothesis'.
Originally and subsenquently, the Lipshutz school argued the intervention a tricoordinate-dianionic copper species based on
product formation. That tricoordinate Cu(I) species was the hypothesis. Penner-Hahn and Huang have argued quite clearly
that spectroscopy and theory 'taken together' yield an alternative hypothesis, albeit an exlusionary one: No species with
three bonds to copper. This is important headway. They likewise point out that the same combo
of theory and measurement point to a dominant species in solution, i.e. bridged 3. This too is hypothesis,
but one entirely consistent with all the data to date. Furthermore, it begins to address the aggregation
problem. Namely, an aggregate of LiCN and [MeCuMe]Li or of Li2CN+ and [MeCuMe]- etc is the primary structural candidate.
Thus, while the details will continue to be refined, two important new points have emerged: No HO and a Gilman-like aggregate
account for the initial species in solution. Important questions that will attract the community's attention for
awhile include: Are there alternative simple aggregates equally consistent with experiment? The IR paper mentioned above explores
this question, though not exhaustively. Are these aggregates large enough? Are there many or few equilibrium partners?
On the latter point, both James and Lipshutz have invoked ESIMS as the basis for believing that cuprate solutions contain a large number
of anionic species. We need to be cautious here. While the technique allows identification of a collection of cationic and anionic masses,
only the cations carry solvent molecules. The anions are 'naked', totally devoid of solvent coordination.
If one accepts James' statement that 'cuprates do not exist as monomers in solution' (a point worthy of critical attention), then one
concludes that solvent has a lot - maybe everything - to do with aggregation as it relates to cuprate chemistry. The complete lack of
solvent for the ESIMS anions at least raises the possibility that the multiplicity of species is an artifact of
the gas phase. This, plus the fact that the ESIMS anions lack a critical ingredient present in
solution (namely an Li+) allows one to tentatively draw the conclusion that ether solution constitution and MS
ion may not have a lot to do with one another. This position does not eliminate the need to ask if there are multiple reactive species
in solution, but it does call for another approach to answer the question. As of now, IR and NMR are most consistent with a single dominant
cluster for Me2CuLi.LiCN. As to the HO question, ESIMS appears not to address it. The species in question
have different numbers of atoms, one consequence of which is that all gas phase ions evaluataed by
ab initio calculations are linear. The situation does raise the amusing question: If one Li+ cation in a constitution such
as R2CuLi.LiCN could be replaced with a non-coordinating cation as in R2CuLi.(R'4N+)CN, would one generate a new reagent with novel
chemical behavior?
The final and provocative point raised by James is the question of reactivity. Are these aggregates we're discussing
critical for it? Nakamura and colleagues suggest they might be (unpublished). Work from our group strongly suggests that the reactivity issue
operates much further down the reaction pathway (JACS 1995, 117, 11023 and 11025). Some very recent NMR work by Bertz and Ogle (unpublished) tends to confirm
this view. Important for the ongoing discussion would seem to be a cleavage between ground state and transition state concepts. Are the ground state deductions about
structure transferable to the rate determining transition state? For normal organics where only a few key bonds
are made and broken, the link is tight and obvious. For organometallics with lots of loose, exchangeable bonds, the link is much
weaker. We should ask hard questions about the connection so as not to be misled by our growing insights into ground state solution
structure.
Bruce H. Lipshutz, accepted 30/Jul/1996
In the course of the discussions on HO cuprates, there seems to be an occasional reference to the recent Bertz contribution in Chem Comm which has led to the belief that there is no difference in reactivity between HO and Gilman cuprates. It must be noted that this study was done, as is usually the case, with cyclohexenone. Unfortunately, this is not a particularly informative educt, since it does not present any barrier (e.g., sterics) with which the cuprates must contend. I surely would have predicted the outcome here as being identical. A more informative comparison would be one where thermal decomposition is not an issue. The second point made by Bertz in his recent paper is that of transmetallation; here, this would not be possible. Only where, say, an alkylation with an iodide is involved would one need to worry about this mode of competition. So what about using something like isophorone at -78°? What about choosing a substrate that contains a stereocenter such that a ratio of diastereomers would result that might be different for the different cuprates being compared? Bertz had the right idea about temperature; in my view, he chose the wrong substrate to gauge reactivity. To make the broad claim that these reagents are of the same reactivity based on one very simple enone...does the Bertz experiment truly merit this conclusion? Brian James will be doing these experiments with isophorone tomorrow (at least that's the plan today). We shall see.
Secondly, there has been reference made to the aggregation state of cuprates, and the electrospray technique as a tool of analysis. The criticism offered is that in the gas phase there is no solvation of anions, and thus it is not obvious that the data reflect true composition. I would counter by saying that there is a fair amount of cationic ESIMS which has been done in solvents other than water, or certainly ethers. Does anyone question this data done in CH2Cl2 or EtOAc? Why is it essential to see solvent aggregates in either the cationic or anionic form to accept the data? Perhaps an interesting experiment that comes out of this discussion is to look at the Ullenius cuprate made in Et2O which has been stripped of solvent and replaced by CH2Cl2. I realize that one molecule of Et2O remains on Li, but we might see something interesting.
© ECHET96. January 1997.