

Binding Studies of Glycosidase Inhibitors with Basic Nitrogen Instead of Oxygen in the Sugar Ring: Modeling of Enzyme-Inhibitor-Complexes
Michael EBNER, Christian E. EKHART* and Arnold E. STÜTZ
Institut für Organische Chemie der Technischen Universität Graz,
Stremayrgasse 16, A-8010 Graz, Austria

Sugar shaped glycosidase inhibitors with basic nitrogen instead of oxygen in the ring have emerged as very important tools for glycobiological research.
Structure activity correlation in this series of compounds have mainly relied on empiric conclusions. Computer aided structure acitivity relationship investigation into inhibitors of D-mannosidases were reported by Winkler and Holan 8 based on the structural similarities of mannosidase inhibitors and comparison of their activities. Only recently crystallographic data on glucoamylase, an exo-glucosidase, have become available and based on these pieces of information, modeling of the active site containing selected inhibitors of different acitivity has become feasable.
We have selected a range of glycosidase inhibitors and have tried to derive data for the estimation of inhibitory acitivity from computer based binding and dissoziation studies of enzyme-inhibitor-complexes
The recently published crystal structure of a glucoamylase1 (.a.-1,4-D-glucan glucohydrolase, EC 3.2.1.3) (3GLY.PDB) as well as the corresponding enzyme-inhibitor-complex2 (1DOG.PDB) with 1-deoxynojirimycin were chosen as model structures. Glucoamylases catalyze the removal of .b.-D-glucose from the nonreducing ends of starch and are widly used in industry for the conversion of starch to glucose syrup. Their modes of action are assumed to follow the generally accepted pathway for the enzymatic hydrolysis of glycosidic bonds as established by Koshland6 and Phillips7.
In context with our ongoing projects concerned with the synthesis and biological evaluation of sugar-shaped glycosidase inhibitors with basic nitrogen instead of oxygen in the ring we wanted to take advantage of the available structural data of the above mentioned enzyme and its complex with the powerful glucosidase inhibitor 1-deoxynojirimycin. In order to gain further insight into the structure-activity-relationships in this class of inhibitors, we have probed the active site with selected derivatives of 1-dexynojirimycin as well as its 5-membered ring analogue 2,5-dideoxy-2,5-imino-D-mannitol. In addition, bicyclic analogues such as castanospermine and australine were chosen for comparison.
All structures were taken either from the Brookhaven National Laboratory Protein Database or the Cambridge Crytallographic Database. The enzyme consists of three functional domains: A catalytic domain (residues 1-440), an O-glycosylated linker domain (residues 411-512) and a starch binding domain (residues 513-616)1

Data and Calculations
All calculations were made using Tripos Sybyl, version 6.2, employing the Tripos forcefield and charges by the Gasteiger-Hueckel-method. Molecular dynamics (MD) runs were made using the Gasteiger-Hueckel charges at a simulation temperature of 300 and 500 K in several intervals of 10,000 ps each. The energy values are given in kcal/mol (Sybyl), the hydrogen bonds of the inhibitors bound to the active site were counted as listed by the sybyl software.

Enzyme-Inhibitor-Complexes
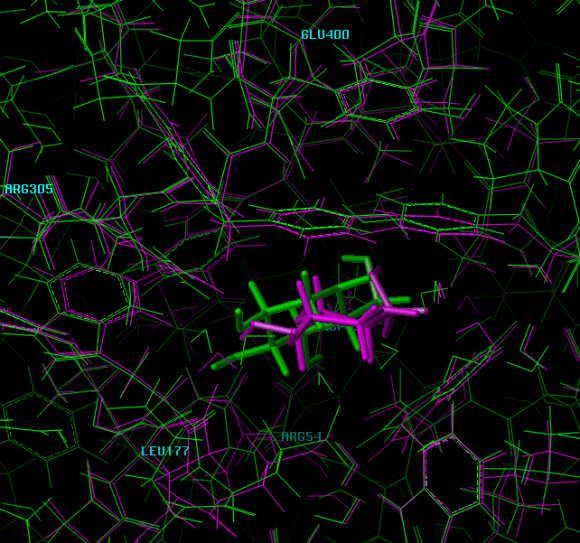
The following inhibitors were modeled into active site of the enzyme-inhibitor-complex using the crystal structure 1DOG.PDB of 1-deoxynojirimycin bound to the active site of glucoamylase.2

300 pix wide |
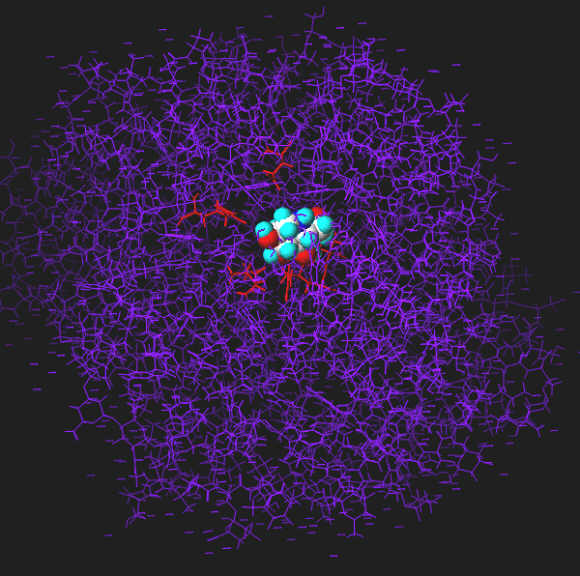
The inhibitor (1-deoxynojirimycin) bound to the active site. The interacting groups of the active site are in red color. Interacting groups and inhibitor are drawn using chapped sticks.
|
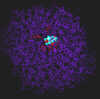
300 pix wide |
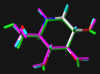
The inhibitor bound to the active site. The interacting groups of the active site are in red color. Interacting groups are drawn using chapped sticks, the inhibitor is shown in spacefill view.
|

300 pix wide |
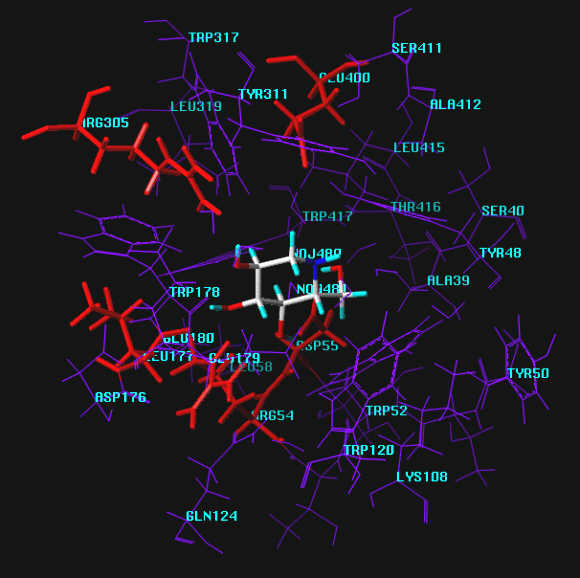
Zoomed view of the active site with groups labeled.
|
Inhibitor Molecules
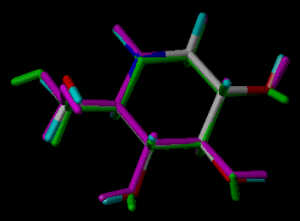
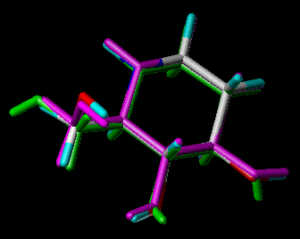
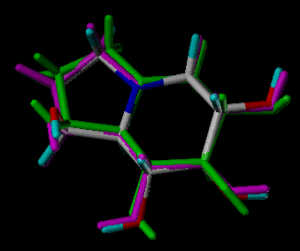
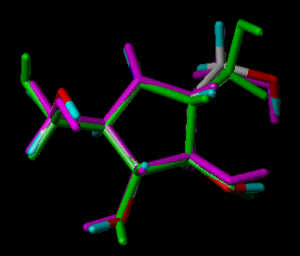
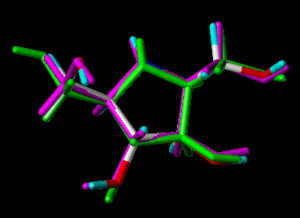
In the following part the inhibitor molecules are presented in three conformations:
green: structure as stored in the Cambridge Crytallographic Database, atom-type-color: conformation of the interaction between enzyme and inhibitor and pink: freely minimized interaction conformation.
|
|
#1
|
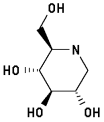
1-Deoxynojirimycin 3
2-Hydroxymethyl-3,4,5-trihydroxypiperidine
| 
MOL1.SKC
| MOL1.PDB, YAXYAX.PDB
, MOL1.MOL2
Energy when bound to enzyme: 18.289
Minimized: 15.825
8 H-bonds, inhibitory activity: 96 uM2
Torsion of bond C5-C6 differs significantly between Cambridge Database and enzyme bound conformation.
| 
300 pix wide |
|
|
#2
|
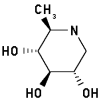
1,6-Dideoxynojirimycin 3
2-Methyl-3,4,5-trihydroxypiperidine
| 
MOL2.SKC
| derived from MOL2.PDB, YAXYAX.PDB
, MOL2.MOL2
Energy when bound to enzyme: 13.210
Minimized: 11.286
7 H-bonds, inhibitory activity: weak (mM range)
| 
300 pix wide |
|
|
#3
|
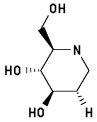
1,2-Dideoxynojirimycin 3
2-Hydroxymethyl-3,4-dihydroxypiperidine
| 
MOL3.SKC
| derived from MOL3.PDB, YAXYAX.PDB
, MOL3.MOL2
Energy when bound to enzyme: 14.154
Minimized: 12.303
7 H-bonds, inhibitory activity: weak (mM range)
| 
300 pix wide |
|
|
#4
|
Castanospermine 3
1,6,7,8-Tetrahydroxy-octahydroindolizine
| 
MOL4.SKC
| MOL4.PDB, BEDELB10.PDB
, MOL4.MOL2
Energy when bound to enzyme: 24.790
Minimized: 20.562
6 H-bonds, inhibitory activity: 8 uM9
| 
300 pix wide |
|
|
#5
|
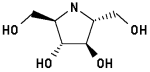
2,5-Dideoxy-2,5-imino-D-mannitol 4
2,5-Dihydroxymethyl-3,4-dihydroxy-pyrrolidine
| 
MOL5.SKC
| MOL5.PDB, HEXPOL.PDB
, MOL4.MOL2
Energy when bound to enzyme: 23.830
Minimized: 19.802
9 H-bonds, inhibitory activity: uM range
| 
300 pix wide |
|
|
#6
|
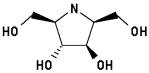
2,5-Dideoxy-2,5-imino-D-glucitol 5
2,5-bis(Hydroxymethyl)-3,4-bis(hydroxy)pyrrolidine
| 
MOL6.SKC
| MOL6.PDB, LEWTOW10.PDB
, MOL6.MOL2
Energy when bound to enzyme: 24.535
Minimized: 20.983
8 H-bonds, inhibitory activity: not determined
| 
300 pix wide |
|
|
#7
|
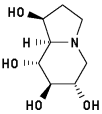
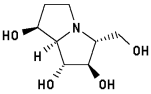
Australine
| 
MOL7.SKC
| derived from MOL7.PDB, HEXPOL.PDB
, MOL7.MOL2
Energy when bound to enzyme: 28.282
Minimized: 24.308
7 H-bonds, inhibitory activity: 6 uM9
| 
300 pix wide |
|
Dissoziation Studies
The pictures are from a MD run of 20,000 ps at 300 K. The view is from outside the enzyme, the inhibitor is slowly moving toward the viewer.
Discussion
In this attempt to draw conclusions from the data obtained by fitting a range of good as well as poor inhibitors into the acive site of glucoamylase emphasis was layed upon the hydrogen bonding interactions between the enzyme and the respective inhibitor. Although this approach disregards the relative basicites of the ring nitrogens interesting correlations between the biological activities and the number of hydrogen bonds in the enzyme inhibitor complexes have been detected.
- 1-Deoxynojirimycin and derivatives
In our approach, 1-deoxynojirimycin, a good inhibitor in the series of 6-membered ring basic sugar analogs, exhibits 8 significant hydrogen bonds to the enzyme probed. The poorly inhibiting derivatives (MOL2 and MOL3) display 7 such interactions each. In case of MOL2 a hydrogen bond between OH6 and Asp55 of the active site, present in MOL1, cannot be formed. The same is the case for MOL3 not being able to interact with a discrete water molecule close to Arg305. Both of these features in the active site appear to be vital for successful interaction.
- 5-membered rings
Of the two 5-membered rings, MOL5 exhibits 9 hydrogen bonds to the active site in additions to all the other interactions found for MOL1 a second hydrogen bond appears to be present between O4 and Arg54. In contrast, MOL6 does not interact with this binding site at all. Instead, an unusual 2-fold interaction of OH3 with the same amino acid was found.
- Bicyclic systems
Remarkably, neither of the two highly potent inhibitors MOL4 and MOL7 of this enzyme showed appreciable hydrogen bonding interactions with the active site in initial calculations (6 and 7 hydrogen bonds, resp.). Taking into account the inherent "meso properties" of MOL7, which can be regarded as an analog of MOL5 with one of the primary hydroxyl groups locked by the second ring, an "upside down" orientation of this compound was investigated subsequently. In this geometry of interaction, MOL7 displays 10 hydrogen bonds, which is the highest number of all molecules probed in this investigation. In addition to all the interactions found for the the orientation looked at first, bonding was found to H2O-500 at Glu400 as well as Arg54. Remarkably, the third additional interaction is formed with Trp178, not found in any other case.
A still enigmatic exception is MOL4, which does not exhibit bonding to H2O-500 at Glu400 at all, a binding site apparently vital for all active inhibitors. It cannot be excluded that this molecule can bind in a different conformation and/or orientation as commonly assumed and displayed in the Cambridge Crytallographic Database. Calculations in this direction are currently persued.

- A. Aleshin, A. Golubev, L.M. Firsov, R.B. Honzatko, J. Biol. Chem., 1992, 267, 19291-19298
- E.M.S. Harris, A. Aleshin, A. Golubev, L.M. Firsov, R.B. Honzatko, Biochemistry, 1993, 32, 1618-1626
- A. Hempel, N. Camerman, D. Mastropaolo, A. Camerman
J. Med. Chem., 1993, 36, 4082
- J. Lamotte-Brasseur, L. Dupont, O. Dideberg, Acta Crystallogr.,Sect.B, 1977,33, 409
- Chi-Huey Wong, L. Provencher, J.A. Porco Junior, Sang-Hun Jung, Yi-Fong Wang, Lihren Chen, Ruo Wang, D.H. Steensma, J. Org. Chem., 1995, 60, 1492
- D.E. Koshland, Biol. Rev., 1953,28, 416-436
- C.C.F. Blake, L.N. Johnson, G.A. Mair, A.T.C. North, D.C. Phillips, V.R. Sharma, Proc. R. Soc. London, Ser. B, 1967, 167, 378-385
- D.A. Winkler, G. Holan, J. Med. Chem., 1989, 32(9), 2084-9
- R.J. Molyneux, Y.T. Pan, J.E. Tropea, M. Benson, G.P. Kaushal, A.D. Elbein, Biochemistry, 1991, 30(41), 9981-7

We appreciate financial support by the Austrian Fonds zur Förderung der Wissenschaftlichen Forschung, Vienna, (Projects 7335, 8415, and 10067).

|

{kind=link}
{kind=link}
{kind=link}