Semi-Empirical Modelling of a Diels-Alder addition catalysed by a cyclic porphyrin trimer
Sylvain Comiti and Henry S. Rzepa
Department of Chemistry, Imperial College, London, SW7 2AY
Introduction
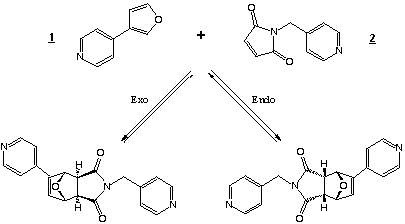
In a recent paper, C. Walter and J. Sander[1] report the catalysis
of the Diels-Alder addition of a furan-based diene (1) and
a maleimide based dienophile (2). They used a porphyrin trimer
catalyst that is inspired by biological enzymes.
The size of the catalyst (285 atoms) is considered beyond that normally modelled using
quantum chemical methods. With advances in the speed of CPU processors and memory, and
the development of methods that scale linearly with size of molecule (MOZYME), we considered
that this reaction could now be successfully modelled
with semi-empirical methods and the results compared with molecular mechanics calculations.
Figure 1 : Diels Alder Addition of the 1 and 2
|
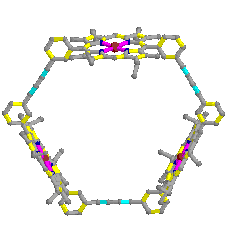
The catalyst we studied is built with a porphyrin trimer motif.
The shape of this trimer is described in figure 2.2.
This has been synthesised[2] and already used for the catalysis
of the reaction of 1 and 2.
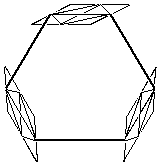
The planes represent the porphyrins and the bold lines represent the links between them. |
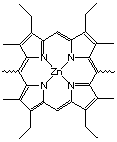 The Zinc-porphyrin
| 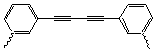
Links
|
Figure 2.2 : Catalyst
Thanks to Dr. James Stewart, the catalyst has been optimized using
semi-empirical calculations : Mopac 93 (H°f = 849.63 kCal/mol, Gradient Norm = 2.48) and Mozyme[4], a new program he is develloping (H°f = 849.69 kCal/mol).
Results
- Molecular Mechanics calculations were carried out using Allinger's MM2 force field parameter, as implemented by the CAChe Mechanics program. Molecule geometry optimizations were computed using the conjugate gradient method.
- Semi-empirical calculations were carried out in the gas phase at the Restricted Hartree-Fock (RHF) level. They were computed on a Silicon Graphics INDY workstation with MOPAC 93.00.
- Without catalyst
Table 1 : Endo/Exo selectivity (without Catalyst)
kCal.mol-1
| H°f(reactants)
|
H°f(products) |
Reaction Enthalpy |
| Semi-Empirical Mopac 93
| PM3 |
endo |
17.76 |
-4.73 |
-22.48 |
| exo | -7.88 |
-25.63 |
Molecular Mechanics
(CAChe)
| MM2 |
endo |
-27.50 |
-13.51 |
13.99 |
| exo |
-11.55 |
15.94 |
kCal.mol-1 |
G°r |
|---|
| Experiments
| endo |
-27.0 |
| exo |
-29.2 |
Table 2 : Experimental results (without catalyst)[3]
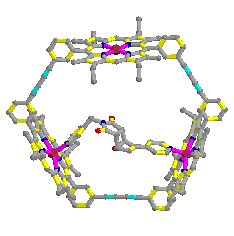
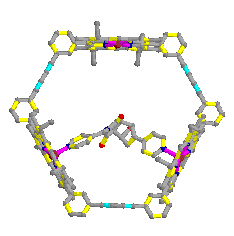
- With catalyst
Table 2.3 : Endo/Exo selectivity (with catalyst)
Molecular Mechanics (MM2)
kCal.mol-1 |
H°f(reactants) |
H°f(products) |
Reaction Enthalpy |
| endo |
-129.00 |
-98.42 |
30.58 |
| exo |
-108.82 |
20.18 |
Table 3 : Endo/Exo selectivity (with Catalyst)
Semi-empirical MOPAC (PM3)
kCal.mol-1 |
G°r |
|---|
| Experiments
| endo |
-27.7 |
| exo |
-28.9 |
Table 2.5 : Experimental results (with catalyst)[3]
Discussion
- The accuracy of the semi-empirical calculations for the substrates alone is
very good (exo-endo energy differences is 1.95 kCal/mol whereas experiments show
that reaction enthalpy is about 2.2 kCal/mol). On the other hand the results for the
catalyst are less accurate. Semi-empirical computations show a higher selectivity
(about 8 kCal/mol) than the true value (about 2.2 kCal/mol). This is probably due to
the fact that the calculations were stopped before the convergence was achieved.
In spite of the same number of cycle (about 150), the endo complex seem to converge
more slowly than the exo (according to the gradients norms).
- Molecular Mechanics calculations results almost fit the semi-empirical results. T
he exo-endo energy differences are 3.15 kCal/mol (without catalyst) and
10.2 kCal/mol (with catalyst), whereas the PM3 energy differences are 1.95 kCal/mol and
8.1 kCal/mol respectively. They tends to be more in favor of the exo
product than semi-empirical calculations.
- Semi-empirical optimizations of large molecular complexes such
as those described above can be made within areasonable amount of
time (about 10 days on a Silicon Graphics INDY R5000 based workstation).
Optimization of transition state and estimation of activation
energies would require a bit more CPU-time, but they are now possible.
In a near future, modelingwill permit an other approach to catalysts activity.
reference
[1]
C. J. Walter , H.L. Anderson and J. K. M. Sanders
Journ. Chem. Soc. Chem. Commun. , 1993 , 458 - 460
[2]
H. L. Anderson and J. K. M. Sanders
Journ. Chem. Soc. Chem. Commun. , 1989 , 1714 - 1715
[3]
C. J. Walter and J. K. M. Sanders
Angew Chem , 1995 , 36 , 217 - 219
[4]
Description of MOZYME
http://iti2.net:80/jstewart/mozdesc.htm