 One novel application of VRML models is in revealing in a simple visual manner
complex structure-activity relationships. Take for example
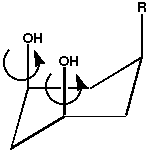
the structures and conformations of a simple molecule such as
1, which can depend greatly on the relative energies of
intramolecular hydrogen bonding between the substituents and the overall
stabilisation of the molecule by solvation.1 These factors in turn depend on
geometrical features such as the axial/equatorial conformation of the
molecule, the relative orientation of the hydroxyl groups to eachother and to the
substituent, and bulk factors such as the polarity of the solvent.
All these effects can be modelled nowadays using quantitative methods based on quantum mechanic
treatment of the molecule, but such modelling produces a lot of data that has to
be interpreted in terms of simple effects.
One novel application of VRML models is in revealing in a simple visual manner
complex structure-activity relationships. Take for example
the structures and conformations of a simple molecule such as
1, which can depend greatly on the relative energies of
intramolecular hydrogen bonding between the substituents and the overall
stabilisation of the molecule by solvation.1 These factors in turn depend on
geometrical features such as the axial/equatorial conformation of the
molecule, the relative orientation of the hydroxyl groups to eachother and to the
substituent, and bulk factors such as the polarity of the solvent.
All these effects can be modelled nowadays using quantitative methods based on quantum mechanic
treatment of the molecule, but such modelling produces a lot of data that has to
be interpreted in terms of simple effects.
To reduce the complexity and enhance any small features from all this data, we
proceeded as follows. Firstly, the energies were calculated as the difference
between heats of formation obtained for a model with a solvent permittivity
(dielectric) e=1 (gas phase) and one with e=80 (water).2 In such a model, we expect an
essentially constant solvation energy will be obtained for molecules where
intra-molecular hydrogen bonding does not alter as a function of geometry, but a
variable solvation energy if intra-molecular hydrogen bonding did vary according
to the conformation of the substituents.
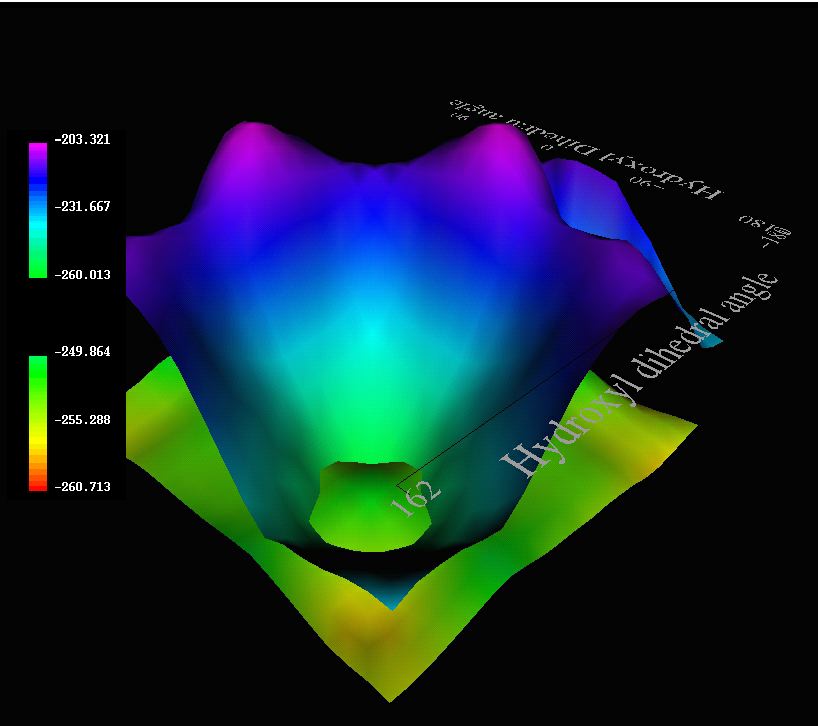
To enable the computed energy differences to be easily interpreted and
associated with individual molecular geometries, the
were presented as VRML models showing a 3D contour map. The dihedral angles of the two
hydroxyl groups were plotted as the X and Y axes and the computed
solvation energy was on the Z axis. The results (PM3 method) for R =
4-methoxyphenyl in the triaxial conformation rapidly revealed to us that
prominent intra-molecular p-facial hydrogen
bonding occurred at geometries associated with two peaks in the
potential surface coded as purple, along with two less prominent peaks
associated with O-H...O interactions. The various regions of the
potential surfaces can have hyperlinks to 3D coordinates, allowing the
reader to explore the geometric features of interest to them simply by clicking with
their mouse button at the feature of interest. The VRML
model also superimposes the tri-equatorial surface (green-yellow
regions), where the lack of any intramolecular hydrogen bonding
renders the surface almost entirely flat. A surface intersection
can be seen, corresponding to a region where the axial and equatorial
isomers are of equal energy.

Substituent effects can be enhanced by subtracting one potential surface from
another (e.g. 1, R=H vs R=4-methoxyphenyl). Here, the prominent purple
ridges now correspond to geometries where hydrogen bonding from the
hydroxyl groups to the p face of the aromatic ring occurs (right).
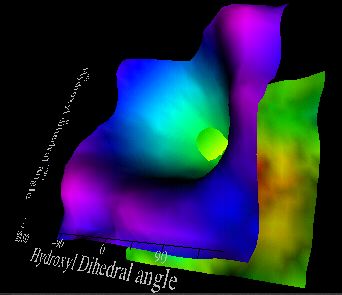 Other unexpected effects can also be discovered using this technique. For
example, altering the the electron demand of the aryl p
face from a donor to an acceptor (R = 4-nitrophenyl) results in significant
stabilisation by oxygen lone pair donation from the hydroxyl oxygen atoms to the
face of the aryl group (left), identified by the different position of the purple
ridge.
Other unexpected effects can also be discovered using this technique. For
example, altering the the electron demand of the aryl p
face from a donor to an acceptor (R = 4-nitrophenyl) results in significant
stabilisation by oxygen lone pair donation from the hydroxyl oxygen atoms to the
face of the aryl group (left), identified by the different position of the purple
ridge.
The advantage of using a VRML model to express the energetics and
geometrical features of such systems is that this allows the reader
to explore the features of interest to them, and not necessarily those
noted and analysed by the original authors.