Summary: An Ab initio Molecular Orbital study using both gas phase and B3LYP/DZVP-COSMO solvation models of the mechanism of palladium insertion into alkyne and aryl carbon-halogen bonds suggests that the mechanism of palladium insertion into alkyne species can proceed via a concerted oxidative addition across the carbon halogen bond. A stepwise mechanism via a s-complex is favoured when a nitro group is introduced onto the alkyne. The palladium insertion into variously substituted aryl fluorides was again found to proceed via a single-step concerted mechanism, and although a s-complex can be located when 2,4-dinitro and 2-nitro substitution is present, the energy of this stepwise route is very similar to the concerted pathway and no clear decision on the pathway can be made. No intermediate s-complex could be located for h6-tricarbonylchromium-complexed fluorobenzene, and only a concerted pathway was identified.
Molecular structures were optimised using either GAMESS18 or Gaussian98,19 the two programs giving identical results at the RHF closed shell level. RB3LYP correlated calculations were done exclusively using Gaussian98. The 3-21G* basis was used for pathfinding calculations, which was then extended to an all electron DZVP20 externally introduced basis set for all atoms. In order to determine the nature of the structures derived from these calculations, calculation of the second derivative matrix was used to verify transition states as having only one imaginary normal mode and equilibrium geometries states as having none. IRC (Intrinsic Reaction Coordinate) calculations were run starting from transition state geometries in both directions, which verified that either 1 or 7 and 3 linked the located transition state. Solvation calculations were carried out with Gaussian 98 using the COSMO model (keyword: "SCRF=(CPCM)") with a permittivity of 78.4 for water as solvent. Geometry optimisation with this keyword set proved poorly convergent for some systems, and the LOOSE keyword was then employed. In some cases, even this relaxation did not facilitate optimisation, as noted in the text. Transition state normal modes and IRC paths were animated by viewing the Gaussian log files using JMol. Coordinates and normal mode animations are available via the supplemental data at http://www.rsc.org/...
Our initial efforts concentrated on establishing the trend across the series Hal=F-I via the alkyne series and showed that the activation energy for concerted oxidative addition via transition state 2 decreases, whilst the exothermicity of reaction increases along the series Hal=F, Cl, Br and I. Insertions across Cl, Br and I are all well established.1 The barrier for Hal=I was very small at the larger basis set level, and no transition state could be located at the smaller 3-21G* level. We also demonstrated that an electron withdrawing group R on the other terminus of the ethyne significantly decreases the barrier and increases the exothermicity of the reaction, whereas the electron donating group R=NH2 has an insignificant effect.
The other major effect along the series Hal=F,Cl,Br,I is that the geometry of the transition states tends towards tetrahedrality. For chlorine the effect is small, but is more apparent for bromine and chlorine (Figure 1c,d). In the RB3LYP DZVP transition state for hal=I, the Pd-P bond of the phosphine trans to the halide is only slightly longer than that of the cis phosphine. As the halide becomes more electronegative, both Pd-P bonds decrease and become less equal in length. Eventually, the longer Pd-P bond corresponds to the cis phosphine and is greatest for Hal=F where the trans Pd-P bond is 2.532Å vs. 2.367Å for the cis Pd-P bond. This can be explained by an increasing degree of pp-bonding interactions between the trans phosphine and the alkyne, which also explains the planar geometry for the Hal=F,Cl transition states compared to the twisted geometry for Hal=Br,I.
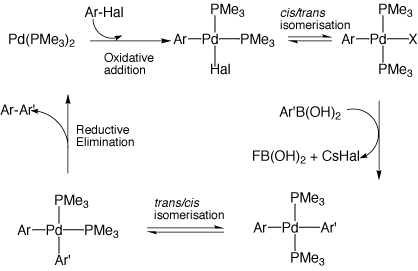
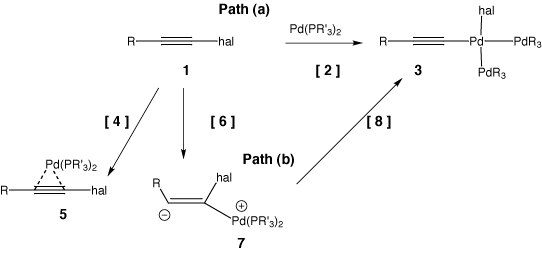
Whereas IRC (Intrinsic reaction coordinate) calculations from the transition state 2, R=H,CN; Hal=F led directly to 1 or 3, that for R=NO2 resulted in the location of an intermediate 7. For this particular case therefore, the stationary point numbered 2 for a gas-phase pathway corresponds to either 6 or 8 on an ionic pathway. This change directly relates to the two possible mechanistic pathways we noted for oxidative addition across a C-halogen bond in scheme 3. Path (a) corresponds to a concerted (non-ionic) oxidative addition across the carbon halogen bond involving transition state 2 whereas path (b) is a stepwise (ionic) reaction involving the formation of an intermediate zwiterionic complex 7 bounded by transition states 6 and 8. The complex 7 is expected to be particularly stabilised by the presence of highly electron withdrawing substituents.
Since significant stabilisation of the zwitterion by solvent would be expected, we applied the RB3LYP//DZVP-COSMO model for evaluating the free energy of solvation, including the free energy corrections for creating the appropriate solvent cavity. Water as solvent was chosen to simulate the maximum reasonable solvation effect. We found that the energies of the gas phase geometry 2 and 7 are lowered by 11.1 and 9.5 kcal/mol respectively when a single point calculation is performed. When the geometry is re-optimised with inclusion of the solvation model, the energy of 7 decreases by a further 3.5 kcal/mol, resulting in a species which is significantly more stable than the reactants. The magnitude of the solvation energy is quite modest for a zwitterionic species; that for the glycine zwitterion for example is 41 kcal/mol at the same level of theory (with additional solvation having its origin in specific hydrogen bonding with solvent). The overall product 3 is similarly stabilised by solvation, whereas the energy of the reactants 1 is essentially unaffected (Table 1).
The geometry of 7 (R=NO2) (Figure 2) reveals that the p-orbitals of the nitro group are coplanar with the carbanionic lone pair and this results in stabilisation through p-interaction . The carbanionic lone pair is also anti-periplanar to the C-F bond, indicating considerable interaction between it and the C-F s antibonding orbital. The geometry at the Pd centre reveals the Pd-C bond to be axial, along with one Pd-P ligand, the other being equatorial. The C-F bond distance of 1.416Å is elongated compared with normal vinyl fluorides (ca 1.28-1.3Å), consistent with the anti-periplanar interaction as noted above weakening this bond as a preliminary to either complete dissociation to form fluoride anion, or of migration to the Pd centre without complete dissociation.
The two transition states (in a solvation model) leading out of the intermediate; 6 corresponding to formation/cleavage of the C-Pd bond and 8 to formation/cleavage of the C-F bond correspond to barriers of 12.2 and 4.5 kcal/mol respectively. Thus with a solvent model applied, transition state 6 replaces the concerted gas phase transition state 2, and the overall barrier to reaction from 1 is now only 1.5 kcal/mol and hence the kinetics are likely to be controlled by entropic rather than enthalpic terms.The potential surface for Pd insertion into aryl halides is analogous to that shown in schemes 2 and 3, with the exception that 5 was not located. Geometries for a series of substituted derivatives are shown in Figure 3 and the energies in Table 2. The RB3LYP/DZVP level was used exclusively for studying these aryl systems. The insertion barriers are similar to those reported for Pt(O) insertions are at a similar level of theory.23. The effect of a single electron-withdrawing group on the activation energy is less for the aryl than for the ethyne species. The calculated B3LYP//DZVP activation energy for fluoroethyne drops by 17.7 kcal/mol when a nitro-substituent is introduced, whereas the aryl analogue is reduced by 5.7 (4-NO2), 13.1 (2-NO2), and an approximately additive 19.4 (2,4-di-NO2) kcal/mol. The most significant effect here is the apparently anomalous result for R=2-NO2, which is attributable to interaction between the nitro group and the Pd centre (cf. Figure 4). The linear geometry of a CN substituent does not allow such an interaction, and here the barrier lowering is very similar for R=4-CN (4.2) and R=2-CN (5.7). We note the similarity of this result to a report of ab initio calculations on aromatic substitution of differently substituted aryl halides22, which revealed that such reactions are predicted to proceed via a single-step mechanism when no electron-withdrawing group is present, albeit with very high energy barriers. The introduction of electron-withdrawing nitro-groups lowered the barrier, but the presence of two nitro groups resulted in a multi-step reaction.
The COSMO solvation model was used to probe whether the stepwise mechanism [path (b), Scheme 3] is energetically viable for the Pd insertion reaction. Calculations at the gas phase geometry revealed quite small solvation-induced changes in the barrier, ranging from -3.8 kcal/mol for 2,4-dinitro, to about +1 kcal/mol for 2 or 4-CN, which implied that an SNAr mechanism (path (b) ) is not viable. However, re-optimisation of the geometry with solvation applied did result in the location of 7 for 2-NO2 and 2,4-NO2 (Table 2). The geometry of this latter species (Figure 4) is noteworthy for the relatively long C-F bond, suggesting a very low barrier for elimination of fluoride anion.
Starting from this optimised geometry, but replacing nitro by cyano and again re-optimising with this substituent lead only to the reactant 1, a repeat of the behaviour found in the alkyne series. This again confirms that pathway (b) is only possible with nitro ring substitution. Location of the two transition states 6 and 8 for this system proved problematic, since the Gaussian COSMO geometry optimisation was found to be unstable and poorly convergent for all controlling parameters investigated. Assuming similar barriers of about 12 and 5 kcal/mol out of 7 as were found for the alkyne system would result in an energy for 6 very similar to the barrier for 2. We conclude that for the nitro-aryl series, the mechanistic pathway (b) would only occur in highly polar solvents, and that it may not constitute a significantly lower reaction pathway than mode (a).
Our final analysis was of reaction of the h6-tricarbonyl chromium fluorobenzene species, a reaction that we have recently reported9,10 occurs in good yields. Houk and co-workers have similarly reported24 that the Cr(CO)3 ligand significantly reduces the barrier towards nucleophilic (anionic) attack at benzene. Our calculated geometry of the transition state 2 reveals the C-F bond to be eclipsed with respect to one Cr-CO group (Figure 5). After the transition state is passed rotation of the Cr(CO)3 group occurs to remove unfavourable steric interactions between the Pd substituent and the Cr(CO)3. Unlike the alkynyl series, the reaction is calculated to be endothermic in the gas phase (Table 2). The difference in the reaction energy between R=Cr(CO)3 and R=2,4-NO2 appears to mostly originate in the Pd-O stabilising interaction in the latter.
The calculated gas phase activation barrier of 40.0 kcal/mol is higher than the activation barrier for insertion into to both 2,4-dinitro fluorobenzene and 4-nitro fluorobenzene. By analogy with the alkynyl series, we anticipate this barrier would be reduced by about 6 kcal/mol if PMe3 ligands replaced PH3), a barrier which is still too high for the facile thermal reaction for this substituent that we observe experimentally. This barrier is reduced by only about 2 kcal/mol when the COSMO model is applied to the gas phase geometry. Geometry optimisation using as a starting point the key parameters obtained for 7, R=2,4-NO2 does not result in the zwitterionic species 7 but rather in reactant 1. Attempts to locate 7, R=Cr(CO)3 starting from either an eclipsed or a staggered orientation of the Cr(CO)3 group also gave this result. We note however that a related species has indeed been isolated from anionic (rather than zwitterionic) addition to Cr(CO)3-benzene.25. This species also has an eclipsing interaction between one Cr-CO bond and the C-H bond at which nucleophilic addition has occurred.
Our failure to locate 7, R=Cr(CO)3 with Pd as nucleophile may relate at least in part to steric factors. Thus the Pd group would have to approach from the aryl face opposite to that coordinated by Cr, and this in turn must force the F atom and its lone pairs into conflict with the eclipsing carbonyl group and its p cloud. A secondary effect is that the h6 coordination of the Cr to the arene ring is significantly reduced (approximately h3, Figure 5), and this is apparently not compensated by the forming C-Pd bond. This problem is lessened for the 2- or 2,4-nitrophenyl system by additional stabilisation of the Pd via O...Pd coordination (Figure 4). Finally we note here that the COSMO model stabilises the product compared to reactant for both R=Cr(CO)3 and R=2,4-NO2, and hence solvation may be the prime thermodynamic driving force for these reactions.
In the aryl series, 2-nitro substitution results in an oxygen-palladium stabilising interaction which additionally helps reduce the insertion barrier. The stepwise and concerted pathways appear more finely balanced than the nitro-alkynyl system, and it is not possible to conclude from the present calculations which mechanistic pathway dominates, if indeed either does. With the fluoro h-tricarbonylchromium benzene system, the formation of an intermediate zwitterion appears sterically hindered, and only a concerted mechanism can be located at this level of theory. However, the predicted barrier for this pathway is rather higher than would be consistent with a reaction that is experimentally observed, and so we cannot exclude the possibility of other mechanistic pathways.
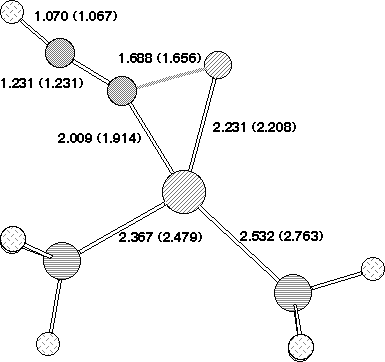
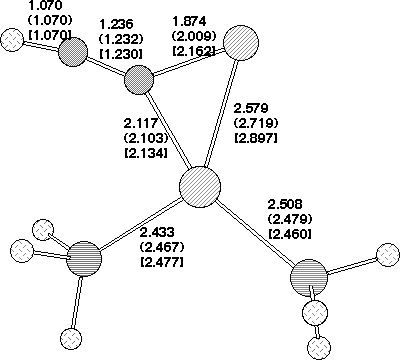
Figure 2. Calculated geometries for stationary point (a) 5 , (b) 2 (c) 3 and (d) 7 on the potential surface for insertion of Pd(PH3)2 into 1, R=O2N, Hal=F Bond length (Å) for B3LYP//DZVP (RHF//DZVP). The geometry of 7 is obtained using the COSMO solvation model.
(a) 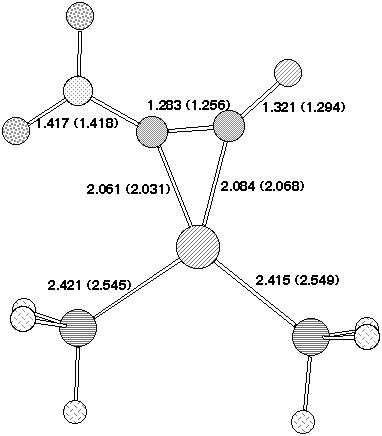
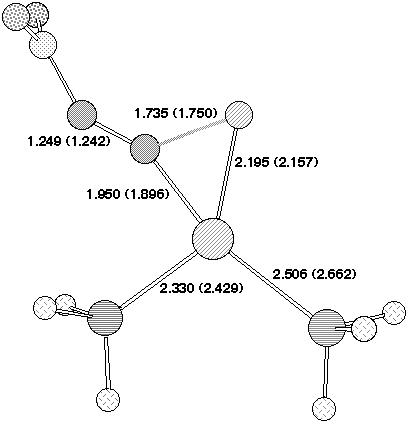
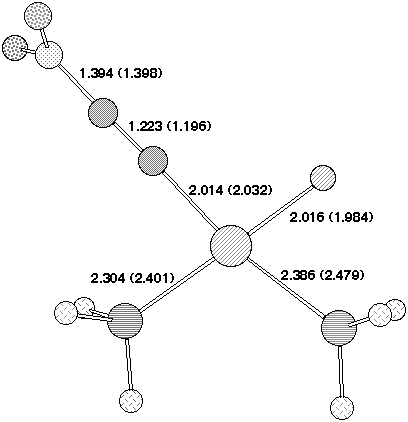
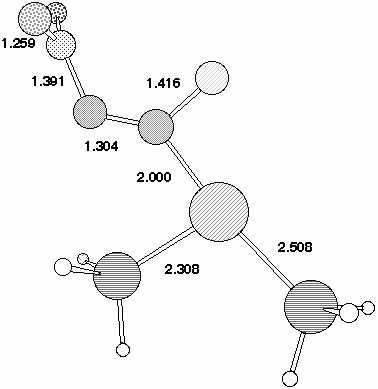
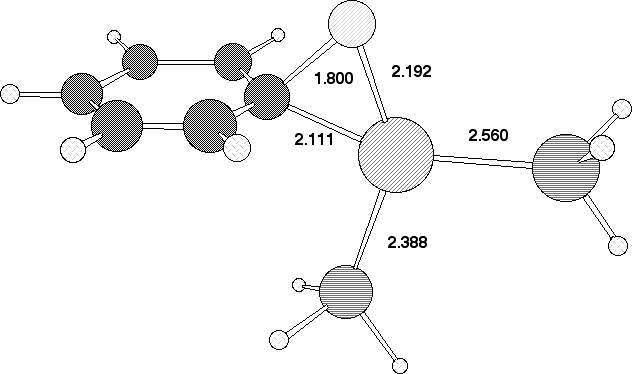
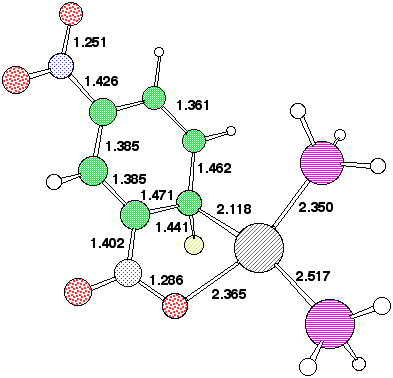
Figure 5. Calculated bond lengths (Å/B3LYP//DZVP)
for the transition state 2 originating from h6 chromium tricarbonyl
fluorobenzene.
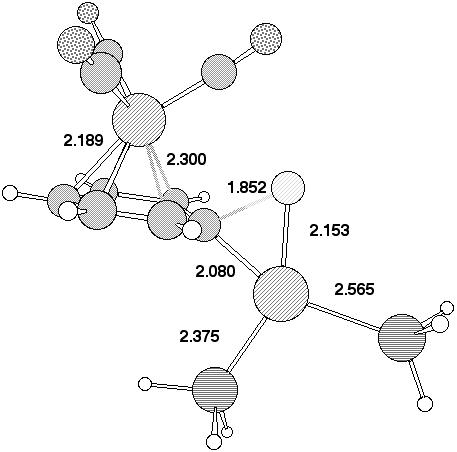
Table 1: Relative Energies, kcal/mol (Total energies given for 1, Hartree) and first order stationary points [transition wavenumber, cm-1] for insertion of Pd(PH3)2 into substituted haloalkynes.
|
R-CC-Hal |
RHF/3-21G* |
RHF/DZVP |
RB3LYP/DZVP |
||||
|
R |
Hal |
Stationary point |
E/DE |
Stationary point |
E/DE |
Stationary point |
E/DE |
|
H |
F |
1 |
1 |
-5797.7672 (0)/-6032.0060a |
1 |
-5802.5714 (0) {6038.5230 (0)}a |
|
|
|
|
2 |
2 |
56.9/[541.0] { (48.2)}a |
2 |
27.8 {21.1}a |
|
|
|
|
3 |
3 |
3 |
|||
|
H |
Cl |
1 |
1 |
1 |
|||
|
|
|
2 |
2 |
2 |
|||
|
|
|
3 |
3 |
3 |
|||
|
H |
Br |
1 |
1 |
1 |
|||
|
|
|
2 |
2 |
2 |
|||
|
|
|
3 |
3 |
3 |
|||
|
H |
I |
1 |
1 |
1 |
|||
|
|
|
2 |
- |
2 |
2 |
||
|
|
|
3 |
3 |
3 |
|||
|
NO2 |
F |
1 |
1 |
1 |
-6007.0821 (0) {-6007.0796 (0)}b |
||
|
|
|
4 |
- |
4 |
4 |
- |
|
|
|
|
5 |
5 |
5 |
|||
|
|
|
6 |
- |
6 |
- |
6 |
{1.5}b |
|
|
|
7 |
- |
7 |
7 |
||
|
|
|
8 |
- |
8 |
- |
8 |
{-6.2}b |
|
|
|
2 |
2 |
2 |
10.10 [328.3] |
||
|
|
|
3 |
3 |
3 |
-16.4 { -30.2}b |
||
|
CN |
F |
1 |
1 |
1 |
|||
|
|
|
5 |
5 |
5 |
|||
|
|
|
2 |
2 |
2 |
|||
|
|
|
3 |
3 |
3 |
|||
|
NH2 |
F |
1 |
1 |
1 |
|||
|
|
|
2 |
2 |
2 |
|||
Table 2: Reltive Energies, kcal/mol (Total energies for 1, Hartree) and transition states 2 [transition wavenumber, cm-1] for insertion of Pd(PH3)2 into aryl fluorides
|
R-aryl-F |
RB3LYP/DZVP |
|||
|
R |
|
E/Hartree, gas phase. |
E/Hartree, COSMO |
|
|
H |
1 |
-5957.5331 |
||
|
|
2 |
-5957.4691 (40.2) |
||
|
Cr(CO)3 |
1 |
-7341.9592 |
||
|
|
7 |
a |
optimises to 1 or 3a |
|
|
|
2 |
40.0 [322.4] |
37.8 |
|
|
|
3 |
[4.7]a |
||
|
2-NO2 |
1 |
-6162.0629 |
||
|
|
7 |
a |
- [22.7 ]a |
|
|
|
2 |
-6162.0159 (29.4) |
||
|
4-NO2 |
1 |
-6162.0738 |
||
|
|
2 |
-6162.0178 (35.1) |
||
|
2,4-NO2 |
1 |
-6366.5892 |
||
|
|
7 |
a |
- [7.4]a |
|
|
|
2 |
18.0 |
||
|
|
3 |
[-4.1]a |
||
|
2-CN |
1 |
-6049.7801 |
||
|
|
7 |
a |
optimises to 1 or 3a |
|
|
|
2 |
-6049.7219 (36.5) |
||
|
4-CN |
1 |
-6049.7861 |
||
|
|
2 |
-6049.7258 (37.8) |
||