Department of Chemistry, Imperial College London, Exhibition Road Campus, London, SW7 2AZ.
High Performance Computing Service, ICT division, Imperial College London, Exhibition Road Campus, London, SW7 2AZ.
This dataset comprises the description of a set of molecular 3D coordinates obtained from quantum mechanical simulation at a variety of theoretical levels, together with associated properties derived from computed wavefunctions for the molecules. These include molecular (harmonic) vibrations, transition state calculations, analysis of the topological properties of the computed wavefunctions (QTAIM) and evaluation of an electron localisation function (ELF) that provides information regarding the local bond properties. The datasets were generated using two quantum chemical computer codes, Gaussian09 and ORCA.
Molecular 3D coordinates, molecular geometry, molecular wavefunctions, harmonic vibrations, transition state geometry, QTAIM topological analysis of electron density, ELF analysis of wavefunction.
This data descriptor aims to provide an overview of datasets which have been made available as a primary component of published scientific articles. It is meant to be "human readable", rather than any formal procedural specification aimed at defining data structures or protocols for generating the data, and is aimed at both those who might wish to generate such datasets and the readers who might wish to (re)-use them. As such, it is not intended to provide a comprehensive review of all the features, but merely to outline the basic features.
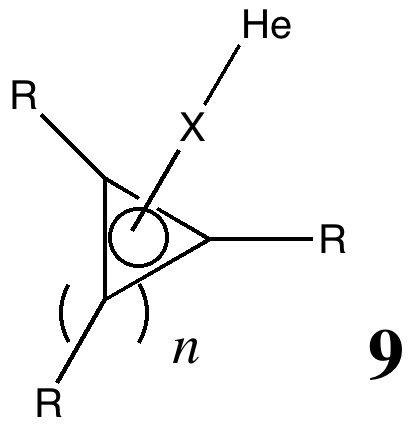
This data set descriptor takes as its scientific context a dataset associated with a published article1 entitled "The rational design of helium bonds", in which the scientific conclusions derive from a sequence of quantum chemical calculations at various theoretical levels. Obtaining molecular coordinates in this manner is a general procedure, applied across the molecular sciences. The scope applies to discrete covalently bound molecules or ionic systems, containing up to around 250 atoms for any atom in the periodic table. The coordinates for these atoms can then be associated with computed wavefunctions for the entire system, which in turn defines the electron density distribution ρ(r) in the molecule. From this distribution, conclusions regarding the length and strength of individual bonds in the molecule can be assessed. In this particular case, it is the strength of bonds to the element helium that are the topic of the article. The computational procedures also allow a potential energy surface for putative reactions of the molecule to be computed, and from this thermodynamic quantities such as activation free energies of reaction ΔG‡298 to be estimated in order to provide estimates of the probable lifetime of the molecule and the rates of its reactions.
Whilst the dataset deployed in the example above is specific to the scientific problem being addressed, the concepts addressed here are in fact quite general to the area of quantum computation in molecular sciences.
The available data relates to the following molecules shown below.
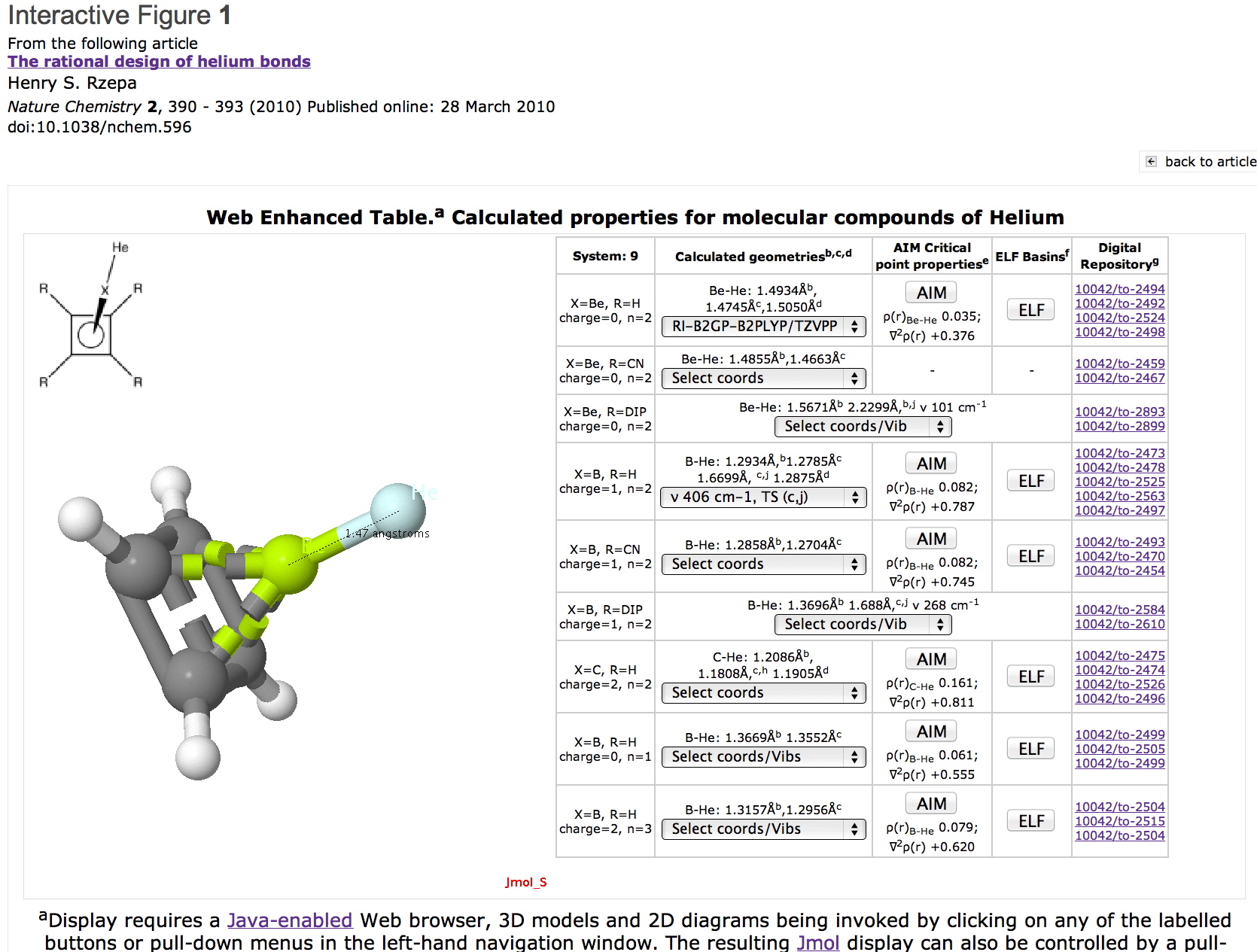
The dataset itself is available2 via the published article in a container therein entitled "Web Enhanced Table. Calculated properties for molecular compounds of Helium". A thumbnail is illustrated in Figure 1.
 .
.
Figure 1. The dataset for quantum computational simulation. Click on graphic to load data.
This table has the following data content:
wget --no-check-certificate https://spectradspace.lib.imperial.ac.uk:8443/dspace/bitstream/10042/$T/2/logfile.out -O $T.log;where the variable in the above example would have the value
$T=to-2899
The dataset is presented to the reader via a Jmol applet9 and is invoked by the use of a Jmol script to provide further annotation of the dataset. Examples of such annotation include:
An example of a script used to annotate is given below:
load "/nchem/journal/v2/n5/media/nchem.596/C4-BeHe-H-ccsdt.cml";zoom 5;moveto 4 0 2 0 90 100; connect (atomno=2) (atomno=4) partial;connect (atomno=5) (atomno=6) single; set measurementUnits Angstroms; measure 2 1; set fontscaling TRUE; font label 24;select atomno=2;label %A Be;select atomno=1;label %A He;
An description of how a dataset can be extracted from its display frame is available.10
There are further examples of the types of dataset as described here.11 There is also discussion of how such datasets can be used to enhance the scientific presentation.12
In this model, the provenance (including date stamp) and authorship of the datasets are formally declared as part of the digital repository entry for each item. Such declarations can also be made in XML-based datasets such as CML as part of the meta-data. These declarations relate to the person in whose name the data was originally generated, and are not necessarily those of the principle investigator or project manager.
The use of job submission and mangement tools provides an opportunity to structure the data capture process from the inception of the calculation. All data required to provide complete provenance of the eventual publication are recorded, ensuring future reproducibility. Furthermore, it enables the output of the calculation to be transparently reprocessed into the various formats useful for publication (CML, etc) without additional effort on the user's part. For all the examples given here, the workflow for generating the datasets is actually formalised in the shape of a job submission portal (Figure 2). This workflow controls submission of a job to the job queuing system for the High Performance Computing resource, and the collection of the job outputs upon completion.
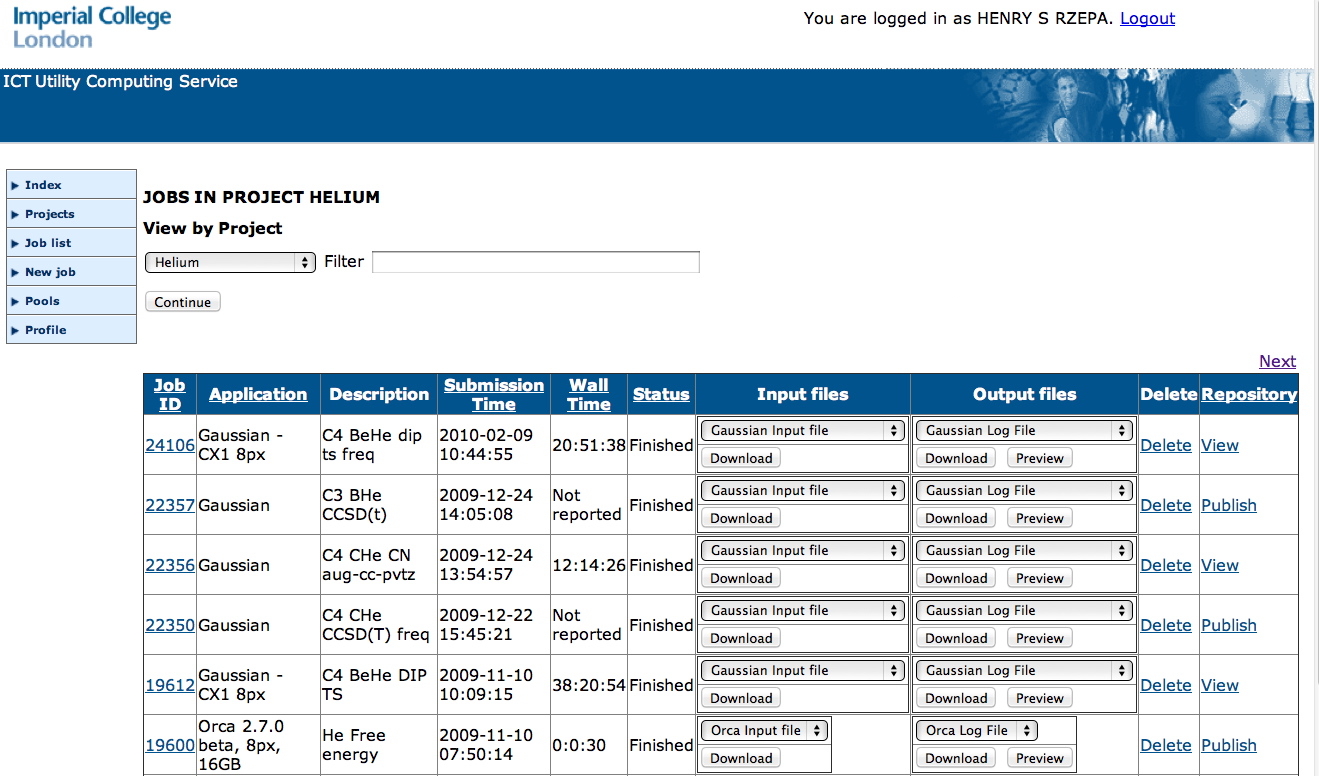
Figure 2. The workflow for data generation.
At this stage, the person who initiated the workflow has the option of publishing these outputs to a digital repository (DSpace for all the examples noted above) at a time of their choosing. An embargo system is used (Figure 3) whereby a declared delay can be configured by the user (up to a maximum of 999 days) if they choose to take no immediate action. After this period is elapsed for any specific entry, it will be automatically published. This component also optionally invokes an RDF declaration (using the FOAF dictionary) of the interests and details of the researcher generating the data. This specification is also deposited in the digital repository, where it can be harvested if needed and which can be used to establish connections with other datasets either created by the same author, or by collaborators of that author.
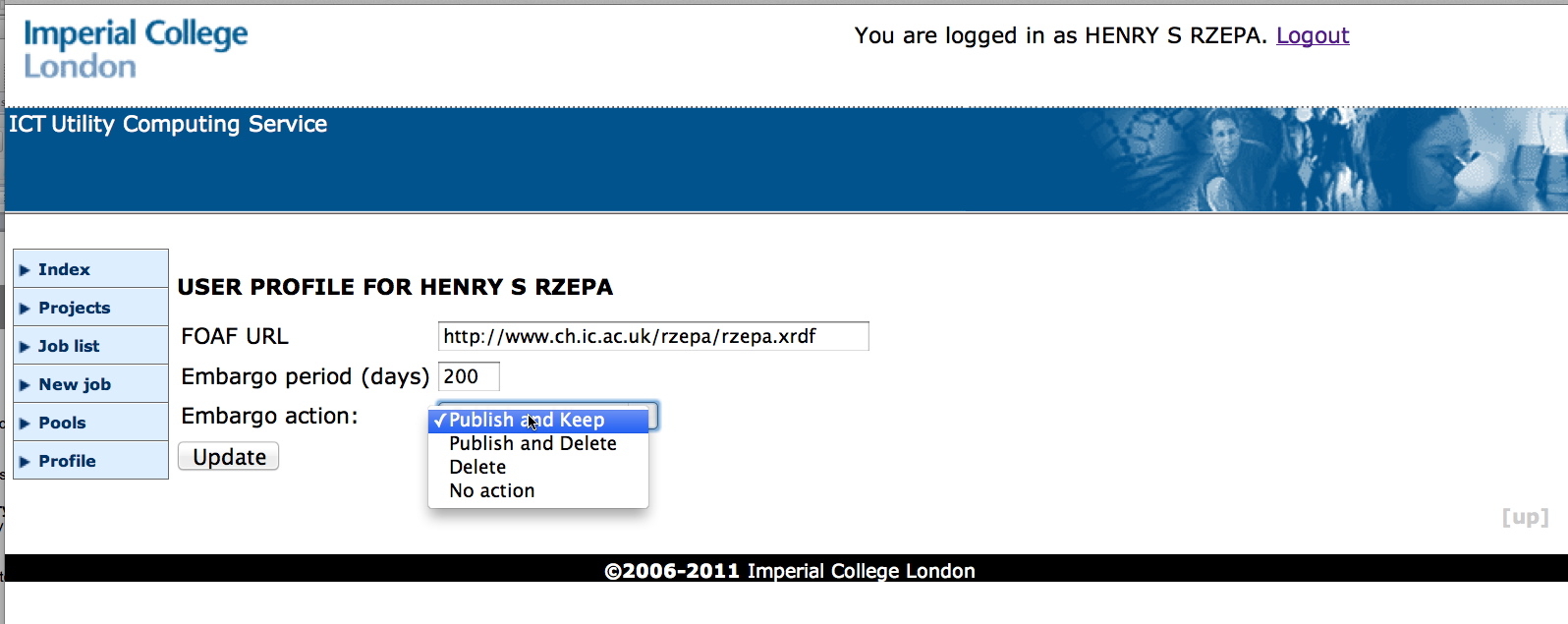
Figure 3. The embargo control.
The actual methodology used to generate the data takes the form of keywords13 driving the Gaussian09 program (equivalent keywords for the other programs6 are available in equivalent form). A thesaurus for mapping such keywords between programs can be generated.6 There is a large measure of consensus on the mathematical descriptions of the procedures used in these programs.
The primary inputs and outputs take the form of an input file for the program in question (Gaussian or ORCA) and the logfile this generates. The input file can be easily assembled using a text editor, although approximate cartesian coordinates for the molecular system being studied will need to be generated. This can be done in several ways
Some further information has to be added to the input file, such as the spin state of the molecule, the charge on the component being calculated and other factors such as the nature of the solvent simulation required, using a a straightforward text editor if need be.
The generated dataset itself is primarily intended to be viewed using either customised closed software such as Gaussview, or a general open package such as Jmol19 (which requires Java 1.3.1). Some forms of this output are in fact "human readable" text files, designed to be inspected using simple text editors. The specialised codes can in fact be used to "round-trip" the dataset, whereby the output can be used as the input in a further cycle, either by the original creator, or by someone who may have acquired the dataset from e.g. the Journal.
The dataset is primarily presented to the reader in the form of a hyperlinked table, as illustrated in the thumbnail above. A mini-tutorial in its use9 outlines the basic procedures involved in re-using data presented by the Jmol applet. A comprehensive set of documentation and tutorials is available.19
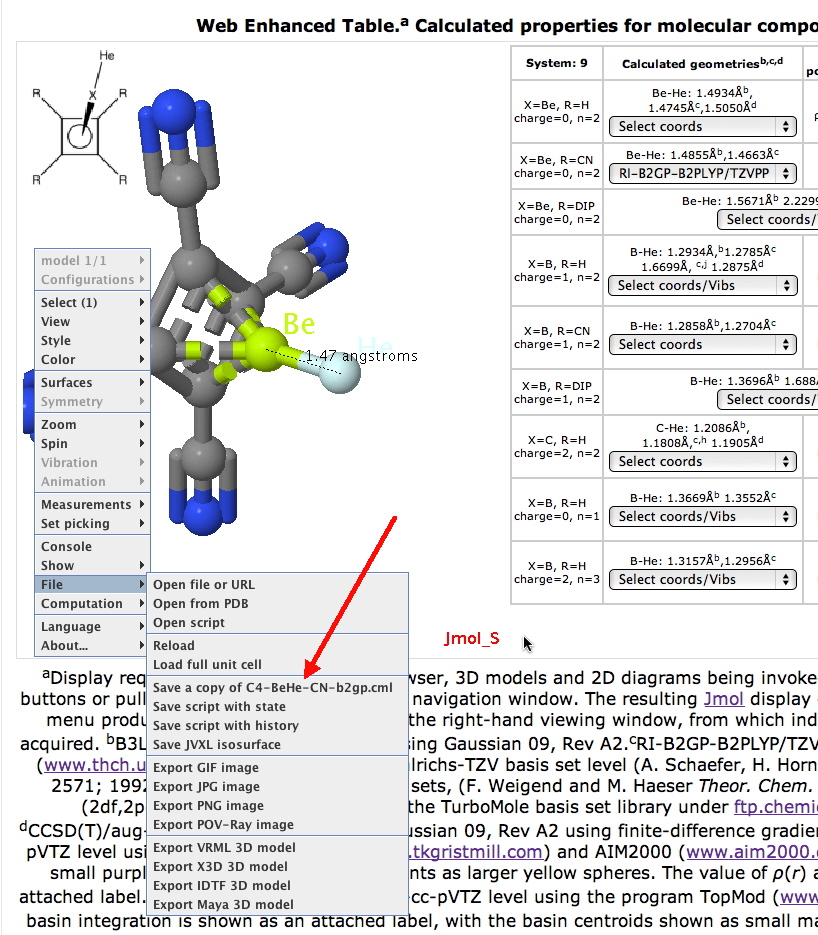
Figure 4.. The Jmol tool, illustrating its usage to obtain data from a dataset.
Figure 4 shows this process in action. The reader loads the dataset they wish to inspect by invoking the appropriate link in the Table. A menu containing further options can be displayed by a right-mouse click in the display window. This is hierarchical in nature; the red arrow points to a file sub-menu which allows various actions. In this case, a request to save a copy of the file to the local file system is being requested. Once saved to the file system, the user can invoke any software tool that supports the file type. Alternatively, the files can be acquired (individually or in volume using automated scripts) from the digital repository for data-mining17 and other operations.
The author has no competing financial interests.
M. J. Harvey and H. S Rzepa, "Data descriptors in molecular science: quantum computational simulation", 2012-07-24. URL:http://www.ch.ic.ac.uk/rzepa/data-descriptors/. Accessed: 2012-07-24. (Archived by WebCite® at http://www.webcitation.org/69OD2TqpJ)