DOI: 10.1039/b002462g

Communication
|
Chemical Communications DOI: 10.1039/b002462g |
Communication |
M÷bius aromatics arising from a
C
|
| Department of Chemistry, Imperial College of Science, Technology and Medicine, London, UK SW7 2AY. E-mail: h.rzepa@ic.ac.uk |
Received (in Cambridge, UK) 28th March 2000 , Accepted 2nd May 2000
Published on the Web 1st June 2000
Replacement of one planar C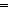 C unit in H³ckel
4n + 2 aromatic rings by a twisted CCC results in
chiral 4n
C unit in H³ckel
4n + 2 aromatic rings by a twisted CCC results in
chiral 4n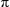 M÷bius aromatic rings.
M÷bius aromatic rings.
electrons, the so-called H³ckel rule of aromaticity.
Heilbronner
1
in 1964 was the first to
suggest that applying a M÷bius twist to the ring would create an
aromatic species if 4n electrons were conjugated. The
M÷bius concept has been widely applied to considering the aromaticity
of pericyclic transition
2
states, but only
recently have candidates for stable M÷bius aromatics been suggested.
Schleyer and coworkers have reported that a M÷bius twisted
conformation of the 4n system
C9H9+ is aromatic
3
on the basis of calculated nucleus independent
chemical shifts (NICS),
2
a technique which
appears reliable and useful at quantifying aromaticity. We recently
reported calculations on two conformations of [16]annulene,
4
one being conventionally antiaromatic, but the
other having a pronounced M÷bius-like twist not associated with any
particular region of the ring. This isomer exhibited a NICS value
consistent with mild aromaticity rather than antiaromaticity. Here we
suggest further candidates for consideration as M÷bius aromatics. Our initial focus was on the chiral species cycloheptatetraene
5. The presence of a CCC substructure in the
7-membered ring reduces the dihedral angle between the 1,3 allene
substituents from 90 to 47¢50░, which has the effect of breaking
the degeneracy of the two highest occupied allene orbitals (Fig. 1; see
ESIå
). Interaction of both resulting
orbitals with the remaining ring orbitals would create a similar
M÷bius topology to that envisaged by Heilbronner, the specific case of
5 resulting in what could be termed a [7]M÷bius annulene. A
NICS calculation (
Our initial focus was on the chiral species cycloheptatetraene
5. The presence of a CCC substructure in the
7-membered ring reduces the dihedral angle between the 1,3 allene
substituents from 90 to 47¢50░, which has the effect of breaking
the degeneracy of the two highest occupied allene orbitals (Fig. 1; see
ESIå
). Interaction of both resulting
orbitals with the remaining ring orbitals would create a similar
M÷bius topology to that envisaged by Heilbronner, the specific case of
5 resulting in what could be termed a [7]M÷bius annulene. A
NICS calculation ( 6.5 ppm, Table
1) establishes that this orbital interaction results in a mildly
aromatic system, being rather less so than benzene itself (NICS 10
ppm). NICS calculations on benzannulated derivatives of 5 were
reported
5
but the aromaticity of the
allene-derived ring was not discussed. The
6.5 ppm, Table
1) establishes that this orbital interaction results in a mildly
aromatic system, being rather less so than benzene itself (NICS 10
ppm). NICS calculations on benzannulated derivatives of 5 were
reported
5
but the aromaticity of the
allene-derived ring was not discussed. The 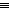 40░ distortion at the
allene unit in 5 is clearly destabilising, since the species
isisolable only at low temperatures, dimerising at higher
temperatures via a 2 + 2
cycloaddition (a process known to be inhibited by bulky groups at the 1,3
positions
6
). The calculated bond lengths for
5 also show some alternation, indicative of a reactive species
(Fig. 2). The HOMO and HOMO 1 AM1
orbits computed for 5 are both symmetric with respect to
C2 axis and derive from the twisted allene system, but
are now delocalised over the entire ring, and show M÷bius topology
(Fig. 2). Heilbronner predicted the HOMO for
a pure M÷bius aromatic would exhibit degeneracy,
1
but purely in a H³ckel MO context. No
degenerate representations in C2 symmetry are necessary
in 5, although we do note that the energy difference between HOMO
and HOMO 1 (1.6eV/AM1) is less than that for allene at this twist
angle (2.2 eV, Fig. 1å
).
40░ distortion at the
allene unit in 5 is clearly destabilising, since the species
isisolable only at low temperatures, dimerising at higher
temperatures via a 2 + 2
cycloaddition (a process known to be inhibited by bulky groups at the 1,3
positions
6
). The calculated bond lengths for
5 also show some alternation, indicative of a reactive species
(Fig. 2). The HOMO and HOMO 1 AM1
orbits computed for 5 are both symmetric with respect to
C2 axis and derive from the twisted allene system, but
are now delocalised over the entire ring, and show M÷bius topology
(Fig. 2). Heilbronner predicted the HOMO for
a pure M÷bius aromatic would exhibit degeneracy,
1
but purely in a H³ckel MO context. No
degenerate representations in C2 symmetry are necessary
in 5, although we do note that the energy difference between HOMO
and HOMO 1 (1.6eV/AM1) is less than that for allene at this twist
angle (2.2 eV, Fig. 1å
). 1 for AM1, Eh
(Hartree) for RHF/6-31G(d)] and NICS values (ppm) for the ground state
structures
1 for AM1, Eh
(Hartree) for RHF/6-31G(d)] and NICS values (ppm) for the ground state
structures
 |

|
|
Fig. 2
Calculated geometry (a) (┼, AM1 (RHF-6-31G(d)) [B3LYP/6-31G(d)]
and form of the AM1 HOMO (b) and HOMO 1 (c) orbitals for
5. |
As a ligand, 5 is unusual in binding metals to both
 faces
faces concurrently (e.g.7),
7
but this is perhaps not unexpected if one
considers it having only a single -face! We also estimate one
consequence of 5 (and its metal complexes) being chiral. Thus a
typical model chiral auxilliary (e.g. R = CHClI or R =
camphorsultamil
8
) results in
diastereoisomers differing in energy by about 0.4¢0.6 kcal
mol1 (AM1), a relatively small discrimination, but
possibly one capable of being increased by suitable design. 5
interconverts with its mirror image via the H³ckel
4n + 2 aromatic 6 (cycloheptatrienylidene),
9
which at the correlated level (CASPT2) has
recently been shown to be the transition state for this process (barrier
20 kcal mol1).Elaborating the theme of substituting CCC for CC
(Table 1) we noted that the small ring
systems 1 and 2 would be classified as antiaromatic;
2 as a conventional 4 antiaromatic H³ckel system with a
triplet ground state and 1 as a 4n + 2 6 M÷bius
antiaromatic system. 1 appears not to exist as a minimum at the
ab initio RHF level, whilst singlet 2 reveals an
antiaromatic (positive) NICS value. No minimum for the anion 3
could be located for this putative 8 M÷bius aromatic, all
optimisations resulting in the aromatic H³ckel valence bond isomer
4, probably because twisting the allene component in 3 to
accommodate a 6-membered ring requires too much energy. This is a lesser
problem in the larger ring monocation 8, which appears to be a
C2 symmetric 8 M÷bius system exhibiting NICS
aromaticity, as is the 8 dication 9. The singlet neutral form
of 9/11 as an antiaromatic 4n + 2 10 annulene
distorts to remove all symmetry and localise the bonds, whereas the triplet
neutral 9/10 retains C2 symmetry as
might be expected of a 4n + 2 excited state M÷bius aromatic,
although the NICS value shows only slight aromaticity (1.3
ppm).
10
Chiral 12 monoanion 10
and the dianion 11 also show aromatic NICS values and have
non-planar twisted geometries (Table 1).
Each ring of the chiral bicyclic analogue of naphthalene 12 shows
only modestly aromatic NICS values. In this instance, the carbene valence
bond isomer 13, a bridged [10] 4n + 2 H³ckel
aromatic annulene, is substantially lower in energy, making it unlikely
that 12 is a viable synthetic target. System 14 is
derived from the novel H³ckel-aromatic S/N systems discovered by Rees
and Surtees.
11
As an 8 M÷bius,
14 has an aromatic NICS value (Table
1). The 12 system 15 has a much smaller NICS value,
which might be related to the larger dihedral angle at the allene termini
(75░, Table 1) reducing the
M÷bius like orbital mixing (c.f. Fig. 1å
). Our results do imply that optimum M÷bius
aromaticity may be achieved at allene twists of
30¢60░.The presence of two or more allene-like chiral M÷bius components
raises the possibility that these can oppose or cooperate. An even number
of the former becomes equivalent to H³ckel; but if the latter the
topological implications become more complex. For 17 two
conformations can indeed be located, one with CS
(H³ckel) and the other with C2 (M÷bius)
symmetry, the latter being lower in energy and higher in aromaticity
(Table 1). In the 10 H³ckel
aromatic conformation of the alkyne 17, the triple bond appears to
act purely as a two electron contributor whereas in the
C2 form, a possible four-electron alkyne contribution
results in 12 M÷bius aromaticity. The alkene analogue 18
has smaller NICS aromaticity values and relative energies, due in part to
the significant non-planarity of 18. More intriguingly the higher
aromaticity of 17 may be due to in-plane trannulene like
aromaticity.
12
The alkyne in 16
appears to act as a M÷bius contributor, cooperating with the allene
component to give a modestly aromatic 12 system. The isomers
19 and 20 also have two conformations. H³ckel
19 as an 8 or 12 4n antiaromatic has the expected
positive NICS value, whilst C2 symmetric 19 is
slightly M÷bius aromatic. Compared to 19, isomers 20
show reversed NICS values (Table 1), and
may be indicative of more complex topological contributions to the
aromaticity. A single M÷bius component inserted into e.g.
[14]trannulene 21 appears much less aromatic than the trannulene
itself (Table 1), but it does conform to
a 4n rather than a 4n + 2 rule for aromaticity. Finally
we note that C2 symmetric 22 has three allene
contributions and as a 12 system it would be expected to be M÷bius
aromatic. Two isomers were identified, one with C2 and
a lower energy and more aromatic form with C3 symmetry
in which all three allene units cooperate.We conclude that a diverse range of M÷bius 4n
aromatic systems can be constructed by using one or more twisted allene
fragments as an initiator. The origins of the M÷bius and H³ckel
contributions to the aromaticity and the nominal electron contributions
(4nvs. 4n + 2) of these systems may be quite
subtle. A dissection of these origins will be reported in a future
article.
concurrently (e.g.7),
7
but this is perhaps not unexpected if one
considers it having only a single -face! We also estimate one
consequence of 5 (and its metal complexes) being chiral. Thus a
typical model chiral auxilliary (e.g. R = CHClI or R =
camphorsultamil
8
) results in
diastereoisomers differing in energy by about 0.4¢0.6 kcal
mol1 (AM1), a relatively small discrimination, but
possibly one capable of being increased by suitable design. 5
interconverts with its mirror image via the H³ckel
4n + 2 aromatic 6 (cycloheptatrienylidene),
9
which at the correlated level (CASPT2) has
recently been shown to be the transition state for this process (barrier
20 kcal mol1).Elaborating the theme of substituting CCC for CC
(Table 1) we noted that the small ring
systems 1 and 2 would be classified as antiaromatic;
2 as a conventional 4 antiaromatic H³ckel system with a
triplet ground state and 1 as a 4n + 2 6 M÷bius
antiaromatic system. 1 appears not to exist as a minimum at the
ab initio RHF level, whilst singlet 2 reveals an
antiaromatic (positive) NICS value. No minimum for the anion 3
could be located for this putative 8 M÷bius aromatic, all
optimisations resulting in the aromatic H³ckel valence bond isomer
4, probably because twisting the allene component in 3 to
accommodate a 6-membered ring requires too much energy. This is a lesser
problem in the larger ring monocation 8, which appears to be a
C2 symmetric 8 M÷bius system exhibiting NICS
aromaticity, as is the 8 dication 9. The singlet neutral form
of 9/11 as an antiaromatic 4n + 2 10 annulene
distorts to remove all symmetry and localise the bonds, whereas the triplet
neutral 9/10 retains C2 symmetry as
might be expected of a 4n + 2 excited state M÷bius aromatic,
although the NICS value shows only slight aromaticity (1.3
ppm).
10
Chiral 12 monoanion 10
and the dianion 11 also show aromatic NICS values and have
non-planar twisted geometries (Table 1).
Each ring of the chiral bicyclic analogue of naphthalene 12 shows
only modestly aromatic NICS values. In this instance, the carbene valence
bond isomer 13, a bridged [10] 4n + 2 H³ckel
aromatic annulene, is substantially lower in energy, making it unlikely
that 12 is a viable synthetic target. System 14 is
derived from the novel H³ckel-aromatic S/N systems discovered by Rees
and Surtees.
11
As an 8 M÷bius,
14 has an aromatic NICS value (Table
1). The 12 system 15 has a much smaller NICS value,
which might be related to the larger dihedral angle at the allene termini
(75░, Table 1) reducing the
M÷bius like orbital mixing (c.f. Fig. 1å
). Our results do imply that optimum M÷bius
aromaticity may be achieved at allene twists of
30¢60░.The presence of two or more allene-like chiral M÷bius components
raises the possibility that these can oppose or cooperate. An even number
of the former becomes equivalent to H³ckel; but if the latter the
topological implications become more complex. For 17 two
conformations can indeed be located, one with CS
(H³ckel) and the other with C2 (M÷bius)
symmetry, the latter being lower in energy and higher in aromaticity
(Table 1). In the 10 H³ckel
aromatic conformation of the alkyne 17, the triple bond appears to
act purely as a two electron contributor whereas in the
C2 form, a possible four-electron alkyne contribution
results in 12 M÷bius aromaticity. The alkene analogue 18
has smaller NICS aromaticity values and relative energies, due in part to
the significant non-planarity of 18. More intriguingly the higher
aromaticity of 17 may be due to in-plane trannulene like
aromaticity.
12
The alkyne in 16
appears to act as a M÷bius contributor, cooperating with the allene
component to give a modestly aromatic 12 system. The isomers
19 and 20 also have two conformations. H³ckel
19 as an 8 or 12 4n antiaromatic has the expected
positive NICS value, whilst C2 symmetric 19 is
slightly M÷bius aromatic. Compared to 19, isomers 20
show reversed NICS values (Table 1), and
may be indicative of more complex topological contributions to the
aromaticity. A single M÷bius component inserted into e.g.
[14]trannulene 21 appears much less aromatic than the trannulene
itself (Table 1), but it does conform to
a 4n rather than a 4n + 2 rule for aromaticity. Finally
we note that C2 symmetric 22 has three allene
contributions and as a 12 system it would be expected to be M÷bius
aromatic. Two isomers were identified, one with C2 and
a lower energy and more aromatic form with C3 symmetry
in which all three allene units cooperate.We conclude that a diverse range of M÷bius 4n
aromatic systems can be constructed by using one or more twisted allene
fragments as an initiator. The origins of the M÷bius and H³ckel
contributions to the aromaticity and the nominal electron contributions
(4nvs. 4n + 2) of these systems may be quite
subtle. A dissection of these origins will be reported in a future
article.Footnotes |
| å Electronic supplementary information (ESI) available: AM1 allene orbitals (Fig. 1), computed 3D coordinates (as PDB files) and selected orbitals (as 3DMF files). See http://www.rsc.org/suppdata/cc/b0/b002462g/ |
| This journal is ® The Royal Society of Chemistry 2000 |
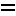 C
C