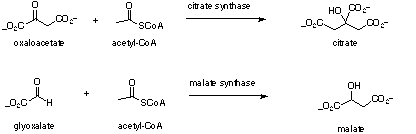
Scheme 1
Published in J. Chem. Soc., Chem. Commun., 1994, 2029.
(c) Royal Society of Chemistry, 1994.
A model for the observed stereochemical course of enzymatically mediated condensations involving fluoroacetyl-CoA is provided by ab initio calculations of the relative stabilities of the E and Z isomers of fluorothioacyl enols and enolates, in which we suggest the enolate intermediate is substantially protonated for citrate synthase but not for malate synthase.
The toxicity of fluoroacetate is attributed to its in vivo activation to fluoroacetyl-CoA and its subsequent conversion to (2R,3R)-fluorocitrate by the action of citrate synthase[1,2]. The accumulation of (2R, 3R)-fluorocitrate inhibits the citric acid cycle enzyme aconitase[3], and citrate transport more generally in cells[4,5], and rapidly induces respiratory failure in mammals. It is an interesting quirk that citrate synthase mediates the formation of (2R,3R)-fluorocitrate, the only toxic stereoisomer of the four possible forms[6]. During the condensation with oxaloacetate (Scheme 1), citrate synthase makes a clear distinction between the two prochiral hydrogens of fluoroacetyl-CoA and abstracts with high selectivity ([[Delta]]G > ~ 3 kcal mol[-1]) only the 2-pro-S hydrogen atom. The condensation proceeds with inversion of configuration at this centre and attack is to the Si-face of the a-carbonyl of oxaloacetate.
Scheme 1
Malate synthase is another example where fluoroacetyl-CoA can replace acetyl-CoA in an enzyme mediated condensation and this system has been studied in some detail[7,8]. For malate synthase both (3R) and (3S) fluoromalates are generated from fluoroacetyl-CoA and glyoxal in a diastereomeric ratio of 4:3. In essence the enzyme displays a slight preference for abstraction of the 2-pro-R over the 2-pro-S hydrogen of fluoroacetyl-CoA. When (2R)-[2-[2]H]-fluoroacetyl-CoA, carrying deuterium in the 2-pro-R position, was incubated with the enzyme then the diastereomeric bias changed to 3:7, now with a preference for 2-pro-S proton abstraction due to an isotope effect. Therefore for malate synthase, unlike citrate synthase, the enzyme is showing a limited ability to orientate the fluorine atom. As part of a more general programme[9] focused on evaluating the stereoelectronic influence of fluorine in enzymatic transformations we became interested in the origin of the diastereoselectivity displayed by citrate synthase with fluoroacetyl-CoA.
It is widely appreciated that fluorine exhibits a limited steric influence over hydrogen in enzyme reactions,[10] and the exclusive stereoselectivity in this case appears unlikely to arise from a steric effect alone. It has been suggested[11] that the selectivity of the citrate synthase reaction may be attributed to a F[....]H hydrogen bond anchoring the fluorine to the enzyme and hence favouring one of the orientations. Such an argument could be extended to malate synthase by invoking a weaker hydrogen bond at the active site of that enzyme. This is however a tenuous argument as fluorine forms only weak hydrogen bonds. A reported screen[12] of crystal structures of organo-fluorine compounds revealed few situations where H[...]F-C hydrogen bonding is obvious. In cases where the F[...]H interaction is apparent the situation is usually highly pre-organised[13]. Interestingly it is more common to find F[...]M alkali metal cation interactions. It may be however that at a geometrically ordered, and desolvated active site, that the strength of a directed hydrogen bond or interaction with an alkali metal cation would be maximal and sufficient to contribute to the overall stability of a particular transition state conformation. This hypothesis therefore remains to be tested.
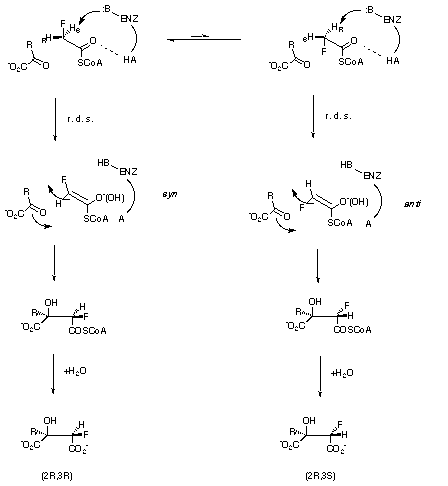
For the two enzymes under discussion, kinetic isotope data (citrate synthase[14] kH/kD = 1.94, malate synthase[15] is kH/kD = 3.9) suggest that the rate limiting step in both cases is proton abstraction from acetyl-SCoA. This is consistent with a much earlier deduction for citrate synthase, that the rate limiting event is the generation of an enolate intermediate[16]. Therefore when fluoroacetyl-CoA is used as a substrate in these enzymes (Scheme 2, R = CH2CO2[-] for citrate synthase, R=H for malate synthase), the prochiral hydrogen discrimination by the enzyme must be related to the relative energies of the syn or anti orientations of the C-F bond with respect to the oxygen atom of the forming neutral enol or enolate anion intermediate, assuming specific binding for the SCoA moiety. We have selected to focus on the energies of the different possible enol/enolate intermediates formed directly after the rate limiting step, with the assumption that these will be directly related to the relative energies of the transition states for proton abstraction.
The relative energies of the two isomers were studied using quantitative molecular orbital theory at the ab initio SCF-MO level, for which reliable calculations for molecules of this size can be made. To enable the use of a large basis set, the SCoA component was modelled with SH. For the anionic enolates, we used a 6-31+(3d) basis set, which includes a diffuse description for the anion. The neutral enol was studied using a 6-31(3d) basis. Correlation energy corrections were included at either the MP2 level or using exchange and correlation density functionals (B-LYP terms), and full geometry optimisation on all species at these levels was performed (Table). Zero-point vibrational corrections did not significantly influence the relative energies at the RHF/6-31(3d) level, and hence were ignored at the computationally more expensive MP2 or BLYP levels. The results show good agreement between the MP2 and B-LYP levels, and clearly indicate that the syn enol is significantly more stable by 4.3 kcal mol[-1] than the trans isomer, an energy difference that is reduced to 1.1 (MP2) or 1.9 (B-LYP) for the anionic enolate system. Both possible orientations of the OH bond in the neutral syn enol were significantly lower than the anti form, suggesting the stabilisation is not due to any intramolecular O-H[...]F hydrogen bond but is rather an example of the "cis" effect as exhibited by e.g. cis difluoroethene[18].
Scheme 2
This study has established that at a high level of theory, fluoroacetate thioesters are deprotonated preferentially to generate syn enols or to a lesser extent syn enolates. A straightforward but striking conclusion to emerge is that the degree of protonation of the enolate modulates the energy difference between the syn and anti geometries. These results therefore suggest a model in which citrate synthase acts not only as a general base in removing the proton, but also as a general acid in protonating the enol[19] to achieve a selectivity of greater than 100:1 in favour of the observed stereoisomer, via a geometry in which the enol adopts the conformation illustrated in Scheme 2 with O and S held as shown.
Our hypothesis extends to malate synthase if it is assumed that this enzyme stabilises an intermediate with greater enolate than enol character. The resultant syn and anti enolates are now much closer in energy and both potential diastereomers of fluoromalate will form. The observed stereochemical preference (4:3) is opposite to that predicted but other minor factors (eg. steric, dipolar/electrostatic interactions, F[...]H-bonding etc.) may contribute and push the bias over in the other direction. An alternative explanation for this diastereomeric mixture is flexibility at the enzyme surface in orientating the C=O and SCoA groups. However such a lack of specificity in binding a co-enzyme-A thioester is contra intuitive and at present we prefer to interpret our results as illustrated in Scheme 2, where O and S remain fixed but that the energy of the syn and anti enolates is similar. We note that these models do not rely on a C-F[...]enzyme hydrogen bond.
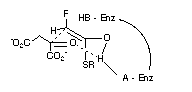
Figure 1
Additionally, our model for citrate synthase helps to define a three dimensional relationship between the general base in the enzyme, the supposed binding site for the SCoA group, and the 2-pro-S hydrogen of the fluoroacetate. A further spatial descriptor, the possible general acid site is thus defined in this model. Additionally this general acid can be strategically placed to protonate the carbonyl of oxaloacetate with the proton contributing to a six membered transition state for the C-C condensation, as illustrated in Figure 1, the next step in the process. It is noteworthy in this respect that citrate synthase will catalyse exchange of the protons of acetyl-CoA when L-malate (but not D) replaces oxaloacetate and that no exchange occurs without L-malate[21]. It has been proposed[22] that a partial conformational change is induced by L-malate binding, and by implication oxaloacetate binding, at the surface of the protein. Clearly L-malate could replace oxaloacetate in Figure 1 and assist the strategic placement of the general acid, through hydrogen bonding, for stabilisation of the developing enol intermediate.
We thank the SERC and the Wolfson Foundation for equipment grants.
The numbering system is that which has developed historically and refers to citric acid as a parent, and is not here altered by the fluorine substituent. See reference 8 for further discussion.
Calculations were performed using the G2 release of Gaussian 92/LDF,[17] with full geometry optimisation using the Eigenvector following method. The final molecular coordinates in chemical MIME format[20 ] are available via the world-wide-web server (www) using the URL
http://www.ch.ic.ac.uk/rzepa/RSC/CC/4_02941K.html, or by clicking here:syn enol anion, syn enol, trans enol anion, trans enol. You will have to configure the MIME types to recognise Gaussian Input files.
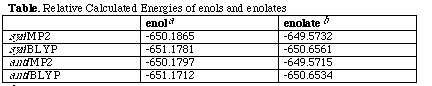
Fluorothioacyl enol. Energies in Hartree at the 6-31G(3d) basis set level with correlation energy corrections at either the MP2 or BLYP level. [b]Fluorothioacyl enolate. Energies in Hartree at the 6-31+G(3d) basis set level.