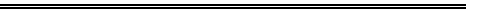
[a]Department of Chemistry, Meston Walk, University of Aberdeen,. AB9, 2UE. [b]Department of Chemistry, Imperial College of Science, Technology and Medicine, London, UK SW7 2AY.
AM1 and PM3 SCF-MO calculations suggest the dimerisation of Hemifullerene
1 to 2 by a mechanism involving six concurrent [pi]2s+[pi]4s
additions corresponds to a stationary point with six negative force constants;
the first stepwise [pi]2s+[pi]4s transition state is found to be highly
unsymmetrical with a large barrier to reaction.
 1
1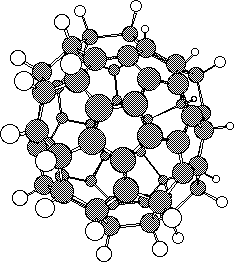 2
2
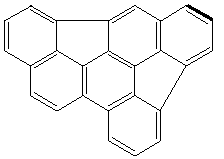 3
3 4
4 5
5
 6
6 7
7
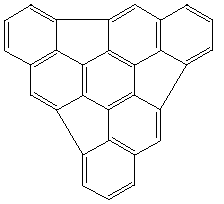
The stationary point corresponding to concurrent six fold [pi]2s+[pi]4s cycloaddition was readily characterised, and was shown to have either 6 (PM3) or 7 (AM1) negative force constants, with a very high energy barrier (Table). The near degeneracy of the first six vibrations indicates that the genuine transition states are likely to correspond to six successive rather than to six concurrent [pi]2s+[pi]4s additions. Our previous finding that metal ions as templates do not appear to enhance the dimerisation, coupled with the present findings that the first [pi]2s+[pi]4s step may have a higher than normal barrier, indicate that high temperatures may still be neccessary for attempts to form the C60 nucleus from 1.
Acknowledgements. We thank the Erasmus scheme for a studentship to SS and the SERC (EPSRC) and Wolfson Foundation for equipment grants.
Theoretical calculations were carried out at the restricted Hartree-Fock level (RHF) AM1 or PM3 semi-empirical methods, as implemented in the MOPAC 93 program.[ ]All structures were optimised using the eigenvector following algorithm, followed by a vibrational analysis to characterise the stationary points.
Computer readable files for Unix, Apple Macintosh or Microsoft Windows systems in MPEG video animation format illustrating the three dimensional properties of these stationary points are available for general access using the world-wide-webb uniform resource locator http://www.ch.ic.ac.uk/rzepa/RSC/CC/4_02038C.html for a period of at least two years from the publication of this paper. A description of how to visualise such material, together with appropriate programs is available from the same sources.
1. M. L. McKee and W. C. Herndon, J. Mol. Struct. (Theochem), 1987, 153, 75; M. J. Plater, Synlett., 1993, 6, 405; R. Faust and K. P. C. Vollhardt, J. Chem. Soc., Chem. Commun., 1993, 1471. See also R. Taylor, G. J. Langley, H. W. Kroto and D. R. M. Walton, Nature, 1993, 336, 728 for formation from naphthalene.
2. M. J. Plater, H. S. Rzepa, F. Stoppa and S. Stossel, J. Chem. Soc., Perkin Trans. 2, 1994, 399.
3. R. Huisgen, Pure Appl. Chem., 1981, 53, 171; A. Firestone, Heterocycles, 1987, 25, 61; W. T. Borden, R. J. Loncharich and K. N. Houk, Ann. Rev. Phys. Chem., 1988, 39, 213; M. J. S. Dewar and C. X. Jie, Acc. Chem. Res., 1992, 25, 537.; K. N. Houk, Y. Li, J. D. Evanseck, Angew. Chemie (Int Ed), 1992, 31, 682. D. A. Hrovat, K. Morokuma, W. T. Borden, J. Am. Chem. Soc., 1994, 116, 1072.
4. F. Bernardi, A. Bottoni, M. J. Field, M. F. Guest, I. H. Hillier, M. A. Robb and A. Venturini, J. Am. Chem. Soc., 1988, 110, 3050; J. H. W. McDouall, M. A. Robb, V. Niazi, F. Bernardi and H. B. Schlegel, J. Am. Chem. Soc., 1987, 109, 4642; M. J. S. Dewar, J. J. P. Stewart and S. Olivella, J. Am. Chem. Soc., 1986, 108, 5771.
5. H. S. Rzepa, P. Molina, M. Alajarin, A. Vidal, Tetrahedron, 1992, 48, 7425.
6. J. W. McIver, Acc. Chem. Res, 1974, 7, 73, J. W. McIver
and A. Komornicki, J. Am. Chem. Soc., 1972, 94, 2625. See also H.
S. Rzepa and W. Wylie, J. Chem. Soc., Perkin Trans. 2, 1991, 939.
Table. Calculated PM3 Enthalpies for the Reaction between 1 and
3-7, in kJ mol[-1].
Structure [Delta]H [Delta]H [Delta]H# r1, r2 (Å) [nu]i
Reactants ts (cm-1)
1+ethene 1135 1309 174 2.06, 2.10 1066
1+butadiene 1198 1357 159 2.10, 2.15 966
1+3 1838 2108 270 1.99, 2.19 1017
1+4 1898 2169 271 1.98, 2.21 1003
1+5 2016 2295 279 1.97, 2.23 985
1+6 2506 2778 272 1.99, 2.22 1003
1+7 2558 2831 273 1.97, 2.25 975
1+1a 2131 2388 257 1.69, 3.03 205
1+1b 2441 2710 268 1.74, 2.91 497
1+1c 2131 3754 1623 2.14, 2.10 e
1+1b,c 2441 4120 1680 2.17, 2.10 f
d 202 312 110 2.14, 2.14 928
[a]
Energies of intermediates corresponding to the product of successive
[pi]2s+[pi]4s additions; 2206, 2121, 2310, 2316, 2189 kJ mol-[1.] [b ]AM1 calculation.[ c
]Stationary point for six concurrent cycloadditions. [d]
Reaction between ethene and butadiene. [e] Values of six
imaginary modes, 1274, 1189, 1189, 1060, 1060, 977 cm[-1].
[f] Values of seven imaginary modes: 1199, 1116, 1116, 982,
982, 904, 78.