This relates to "one-dimensional" spectra - for 2-D and higher spectra it
may be better to use NETCDF as a NOTATION within a CML file
CML holds spectra as an XLIST with two ARRAYs (one for X - frequency, wavelength,
etc and the other for Y - absorption, intensity, etc.). Mass spectra have already
been shown in the tutorial. The following
are examples of files read directly in with a command of the form:
java pmr.cml.JCAMP mynmrfile.jdxwhich uses the JCAMP parser of CML.
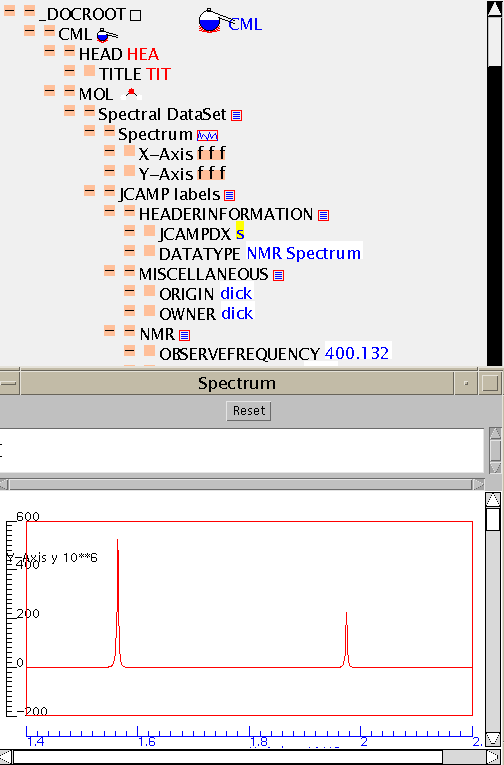
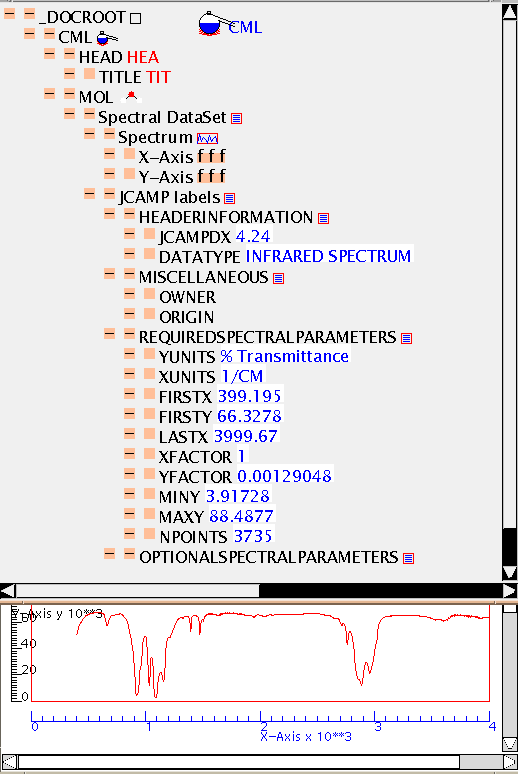
There is no fundamental difference between an NMR spectrum amd an IR one, and the JCAMP parser manages both. Unfortunately the JCAMP spec does not have a field specifying the chemical shift, so all files are either represented in frequencies, or the user has to find some way of adding the chemical shift offset. The IR spectrum is rendered in whatever units (frequency or wavelength) the spectrometer outputs.
The is a small problem with display in that it is often conventional for the spectrum to run 'backwards' with high values on the left. (Of course this doesn't affect the values of the data.) Since CML is a very general tool, it will normally present all numeric data in a consistent manner to the application program. If the author wants to display data in an unconventional way, they can do this with the DISPLAY attribute. Alternatively it may be appropriate to devise special application software that is only used on certain types of data, or can recognise what sort it is.
Here is the raw cml file (ir.cml). The bulk of the spectral values have been omitted for clarity.
This is the IR spectrum of the compound in the tutorial.
Here is the raw cml file (nmr.cml). The bulk of the spectral values have been omitted for clarity.
This is the NMR spectrum of the compound in the tutorial.