Nicolaou et al adopted a convergent approach, the key bond disconnections of which are summarised below:-
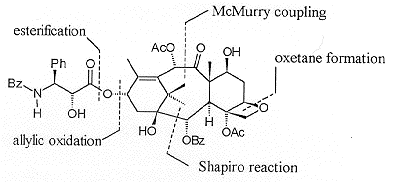
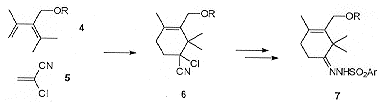
The proposed disconnections of the central 'B' ring leads to a simplified nucleophilic 'A' ring fragment and an electrophilic 'C' ring fragment.The 'A' ring fragment (Scheme 1) was synthesised via the high-yielding diels-alder reaction of diene 4 and 1-chloroacrylonitrile 5 to give the cycloadduct 6 which was then readily converted to the related hydrazone 7.
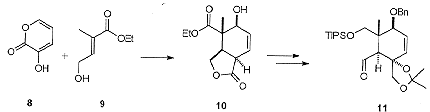
The 'C' ring fragment was similarly prepared via a Diels-Alder strategy (Scheme 2). However, to control the regiochemistry of the cycloaddition it was necessary to tether the diene 8 and dienophile 9 as the boronate. The cycloaddition then proceeds smoothly, and following decomplexation gave the rearranged bicyclic lactone 10 in good yield. Compound 10 was converted over several steps to the required aldehyde 11.
Conversion of 7 to the nucleophilic vinyl lithium species using the Shapiro reaction and coupling with 11 proceeded smoothly to give the required product as the C2-a hydroxy isomer. The stereochemistry at C2 was controlled by the preferential attack of the vinyl lithium species on the re-face of 11. The C1 hydroxyl was then introduced by directed epoxidation of the C1, C14 double bond, followed by LiAlH4-mediated reductive cleavage. The resulting diol was then protected as the cyclic carbonate to give compound 12 (Scheme 3), arranging the substrate for cyclisation. Unmasking the latent aldehyde functionality at C9 and C10 gave the dialdehyde 13.
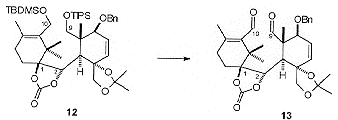
Ring closure of 13 was effected by a McMurry-type cyclisation, giving the diol 14 (Figure 2) in an optimised yield of 23%. A number of other products, resulting from rearrangement of the initially formed oxygen radicals, were also isolated.
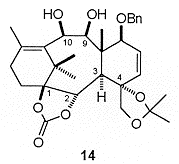
At this stage a resolution was carried out giving the enantiomerically pure diol with the natural configuration in 74% yield. Selective acetylation of the more reactive C10 hydroxyl followed by oxidation at C9 gave the hydroxyl ketone 15 (Scheme 4). Hydroboration of the C5, C6 olefin was problematic resulting in a mixture of the required C5 alcohol and the C6 isomer. At this stage the removal of the acetal was necessary to aid separation resulting in the isolation of the triol 16.
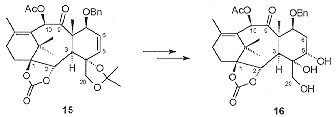
Degradation studies revealed that it was necessary to replace the C7 benzyl ether with a silyl ether prior to oxetane formation. Thus 16 was deprotected and reprotected as the triethylsilyl ether. Monosilylation of the C20 hydroxyl and activation of the C5 hydroxyl as its triflate followed by the mild acid treatment resulted in the isolation of the oxetane containing 17 (Scheme 5). Acetylation of the C4 hydroxyl followed by the selective opening of the cyclic carbonate with PhLi revealed the benzoate moiety.
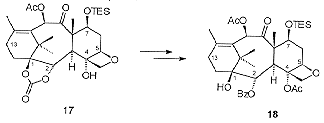
The C13 oxygenation was introduced by a chromium mediated allylic oxidation of 18 followed by stereospecific reduction of the resulting enone. Coupling with a b-lactam side chain derivative followed by desiylation gave Taxol® in a yield of c.a 0.01% from commercially available materials.
|
Glossary Bz = Benzoyl ; Ac = Acetyl ; Bn = Benzyl ; TIPS = Triisopropylsilyl ; TBS or TBDMS = tert-butyldimethylsilane ; TES = triethylsilyl ; BOM = benzyloxymethyl ; TMS = trimethylsilyl ; Tf or triflate = Trifluoromethanesulfomate
|
REFERENCES
Nicolaou, K.C.. J. Am. Chem. Soc. 1995, 117, 624-659
Nicolaou, K.C. Nature. 1994, 367, 630-634
Nicolaou, K.C : J.Chem.Soc. Chem.C ommun. (1994) 295
Nicolaou, K.C. J. Am. Chem. Soc. 1997, 119, 7960-7973
![]()