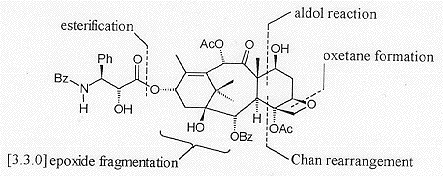
Holton's approach is linear in nature, with a key step being the fragmentation of a highly strained bicyclo [3.3.0] system to form the 'AB' ring carbon skeleton. The C and D rings are then added sequentially (Figure 3).
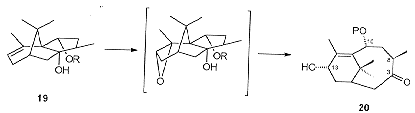
The synthesis starts from (-)-camphor which is converted to the bicyclo [3.3.0] system 19, a substituted analogue of b-patchouline. Epoxidation of 19 followed by an in situe rearrangement leads to 20 (Scheme 6) which contains the complete homochiral 'A' ring, together with all the necessary methyl groups and oxygen functionality in both the lower and upper regions suitably placed for further modification.
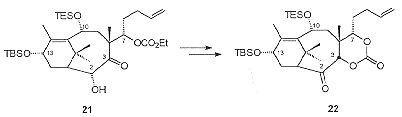
A diastereoselective aldol reaction of 20 with 4-pentenal, protecting the resulting C7 hydroxyl as a carbonate, was followed by hydroxylation at C2 to give the hydroxyl carbonate 21 (Scheme 7). red-Al-mediated reduction of 21 gave a C3, C7 diol which was protected as a cyclic carbonate. Oxidation of the C2 hydroxyl then produced 22.
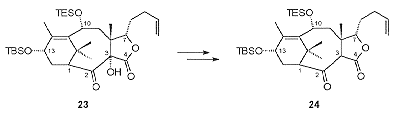
Treatment of 22 with base resulted in an unusual stereo selective Chan rearrangement to give the lactone 23 (Scheme 8). Removal of the C3 hydroxyl by a SmI2-mediated reductive cleavage resulted in the formation of the stable C2 enol which, upon treatment with silica gel, was converted to the required cis -fused lactone 24.
Treatment of 24 with excess LTMP effected deprotonation at C1, which facilitated the introduction of the C1-b hydroxyl. The deprotonation at C1 is of note as the proton at C3 would normally be expected to be more acidic. Directed reduction of the C2 ketone revealed the C1, C2 diol which was subsequently protected as a cyclic carbonate 25 (Scheme 9). Oxidative degradation of the vinyl side chain followed by Dieckmann cyclisation revealed the required 'ABC' ring skeleton 26.
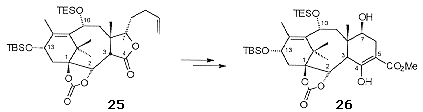
Cleavage of the C5 carbomethoxy group with phenylsulfide gave the required C4 ketone. The oxygenation at C5 was introduced by oxidation of the enolate of the C4 ketone, trapping the intermediate in situ with trimethylsilyl chloride to give 27 (Scheme 10). Methylenation of 27 followed by dihydroxylation allowed the isolation of the diol intermediate which on activation of the hydroxyl as the mesylate underwent base-mediated cyclisation to give the required oxetane 28.
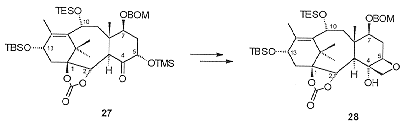
Acetylation of the C4 hydroxyl proved difficult, presumably due to the steric bulk present on the a-face of the molecule, proceeding in only 70-75% yield. This was followed by desilylation of the C10 triethylsilyl ether and selective opening of the cyclic carbonate with PhLi to give 29 (Scheme 11). The oxidation at C9 was introduced by benzenselenic anhydride oxidation of the C10 ketone followed by rearrangement to give 30.
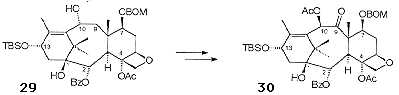
Deprotection of the C13 silyl ether, in common with the difficulties observed with the C4 acetylation, was problematic. However, it was found that the use of TASF resulted in the required desilylation in 94% yield.
Coupling of the resulting alcohol with a b-lactam followed by further desilylation gave Taxol® in a yield of 0.1% from commercially available materials.
|
Glossary Bz = Benzoyl ; Ac = Acetyl ; Bn = Benzyl ; TIPS = Triisopropylsilyl ; TBS or TBDMS = tert-butyldimethylsilane ; TES = triethylsilyl ; BOM = benzyloxymethyl ; TMS = trimethylsilyl ; Tf or triflate = Trifluoromethanesulfomate
|
REFERENCES
Holton, R.A. J. Am. Chem. Soc. 1994, 116, 1597-1598
Holton, R.A. J. Am. Chem. Soc. 1988,
110, 6558-6560
![]()