Brown's famous paper describing the formation and uses of alkyl boranes includes mention of the carbonylation of such species (DOI: 10.1021/ja00997a044) resulting in the formation of ketones.
At least three possible mechanisms can be envisioned (a-c). The step common to all three is TS1, which represents the migration of one alkyl group (R') from boron to carbon.
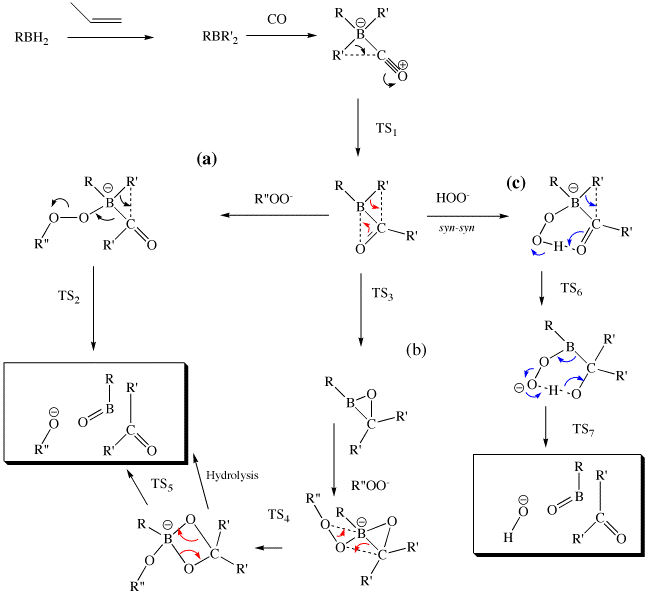
The computed transition states (at the B3LYP/6-31G(d) level for all processes reveal the following:
In pathway (a), TS1 is followed by reaction with the R"-O-O- reagent to give a tetrahedral boron intermediate, followed by TS2, the transition state for the migration of the second group R', also to carbon, but this time accompanied by fragmentations resulting in elimination of both R-B=O and of R"-O-.
This is closely related initially to pathway (a), but the stereochemistry is now syn-syn. This results in an intermolecular hydrogen bond, which develops into a full blown proton transfer during reaction (TS 6, R"=H only) , COI: 10042/to-401, to produce a tetrahedral intermediate , COI: 10042/to-404 which can then undergo an ene-like pericyclic reaction (TS7 , COI: 10042/to-403) to give the final products.
| TS1 (R=R'=R"= Me) [-257.8992] COI: 10042/to-397a |
TS2 (R=R'=R"= Me) [-448.2084] COI (anti) 10042/to-396 (syn1) 10042/to-395 (syn2) 10042/to-400 |
|---|---|
aCOI (Chemical object identifer) points to the entry in the digital chemical repository.
An alternative mechanism rearranges the timing of the bond formations/cleavage, in which the second group R' migrates before the hydroperoxide attacks the boron centre (TS3) to form an unusual 3-membered ring intermediate containing one each of C, O and B. Only then does the hydroproxide coordinate to the B, to form a further intermediate which can then ring expand (TS4 ) to a 4-membered ring ( ). The resulting borinate ester can then be hydrolised to form a C(OH)2 acetal, which eliminates water to form the final ketone product, or which alternatively can underto a 2+2 cyclo-elimination (TS5 COI: 10042/to-402) to give the same products as before.
| TS3 (R=R'=R"= Me) {-190.2157} [-257.8455] COI: 10042/to-398a |
TS4 (R=R'=R"= Me) [-448.2444] COI 10042/to-399 |
|---|---|
The relative energies for all the transition states reveal that the rate determining step is in fact TS1, and that of the subsequent pathways, the "lowest high point" is TS7, and hence that (c) edges it over (a), with (b) nowhere in sight.
| System | Energy, Hartree | ΔΔG, kcal/mol |
|---|---|---|
| Reactant | -257.834192 -150.896296 = 408.730488 | 0.0 |
| TS1 | -257.807228 -150.896296 = 408.70354 | 16.9 |
| TS1 Product | -257.835003 -150.896296 = 408.73129 | -0.5 |
| TS2 | -408.789490 | -37.0 |
| TS3 | -257.753396 -150.89630 = 408.64969 | 50.7 |
| TS3 Product | -257.843650 -150.89630 = 408.73994 | -5.9 |
| TS4 | -408.820527 | -56.5 |
| TS5 | -333.085676 -75.729285 = 408.81496 | -53.0 |
| TS6 | -408.81244 | -51.4 |
| TS7 | -408.79947 | -43.3 |
| Final Product | -408.93600. | -128.9 |