Practice Problems in Pericyclic Reactions.
The following are a set of practice problems in Pericyclic Mechanisms,
prepared by Henry Rzepa for the for a second year course in Pericyclic Reactions at the
Department of Chemistry, Imperial College.
Each of these problems is intended to take between 1-3 hours to solve.
They are NOT typical of examination questions! Please note also that the compound numbers are not consecutive between questions.
Qu 21
Back to Problems
Qu 22
Qu 23
Qu 24
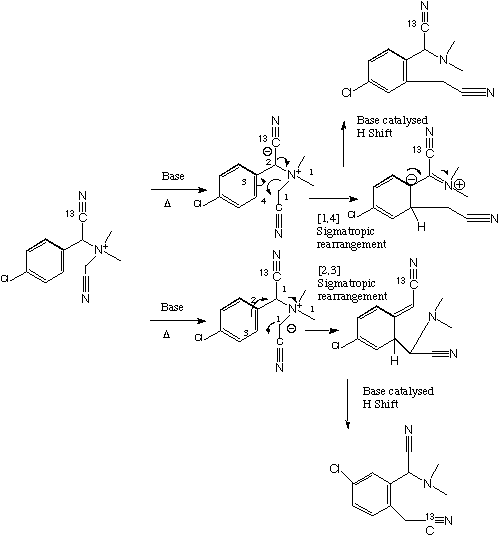
Various cycloaddition or electrocyclisation reactions are possible, followed by either acid/base catalysed [1,3] H shifts (these are not possible as pericyclic reactions) or just possibly [1,7] H sigmatropic shifts. The original literature citation is Tetrahedron, 1992, 48, 7425.
Back to Problems
Qu 25
The reduction in 1H NMR vinyl protons ( d 5.0 - 5.8) from 8 in the reactant C to 4 in the intermediate X suggests a p2s+p4s Diels-Alder type cycloaddition reaction involving a diene and an alkene component. The strongly non-equivalent methylene protons adjacent to O ( d 3.85, 4.15) implies the adjacent carbon is chiral. If the diene component is taken as part of the 6-membered ring, then either of the double bonds in the butadiene sidechain can act as the dienophile, each addition having two different regiochemistries. Of these four possible products, only two have precisely 4 vinylic protons and an oxy-methylene next to a chiral centre, i.e. X and one isomer.
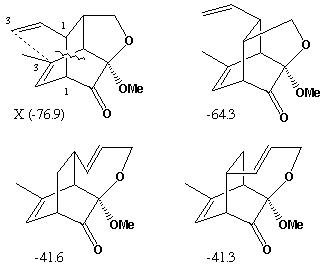
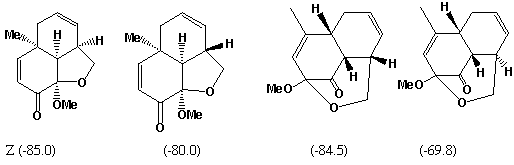
The other possibility is that the sidechain acts as the diene and either of the double bonds in the 6-ring acts as the dieneophile. One addition mode leads directly to Z itself (we assume that Z and X are different!). The other addition mode would give rise to species with only 3 vinylic protons,
The compound shown as X can undergo a simple [3,3] sigmatropic shift to give Z directly. The other isomer compatible with the NMR data cannot be converted to Z. The stereochemistries of the Diels-Alder additions all result from suprafacial addition across both the diene and the alkene, as required by the selection rules, whilst a suprafacial migration in the [3,3] rearrangement gives the stereochemistry shown for Z.
Molecular Modelling. Use of a modelling method such as PM3 (semi-empirical molecular orbital) allows relative energies for all the putative species involved to be calculated. That for compound C is calculated as -49.3 (kcal/mol), with the corresponding values for each possible intermediate and product being shown against the structure above. As can be seen, the most negative (most stable) corresponds to the intermediate X, whilst the most stable product is also the one actually formed. These relative energies are associated with the minimisation of bond angle and torsion strains in the cyclic systems. Models for all these species can be inspected here
Literature Reference : R. Carlini, K. Higgs, C. Older, S. Randhawa, and R. Rodrigo , J. Org. Chem ., 1997, 62, 2330.
Qu 26
L might be expected to form with the two oxylithium centres anti, to minimise charge repulsion and to accommodate the lithium counterion solvated with THF. Electrocyclic ring opening (accelerated by the oxyanion characteristics) proceeds with conrotation (4n electrons, Mobius transition state) to give M, again with the two oxylithium centres presumed on the "outside" to minimise charge repulsion (torquoselectivity). Electrocyclic ring closure conrotation (4n electrons, Mobius transition state) proceeds with an antarafacial component to give predominantly trans stereochemistry at the newly formed C-C bond. Protonation of one enol centre gives the more stable cis 5-ring fusion. The final product is formed by a simple aldol condensation from N, again in a manner which gives a cis ring fusion. Molecular modelling reveals this stereoisomer to be significantly more stable than the trans fused form. Interestingly, the mono-anion of N can exist in two conformations, with the carbonyl group oriented either syn or anti to the adjacent fused 5-ring. The syn ("up") form is the one that leads to the final observed stereochemistry of the resulting alcohol, although it is actually slightly less stable than the anti isomer. This suggests that aldol formation is a reversible process driven by the thermodynamic stability of the final product.
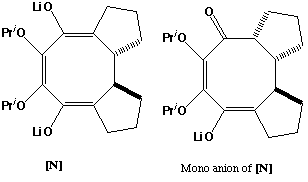
Literature Reference: L. A. Paquette and T. M. Morwich, J. Am. Chem. Soc ., 1997, 119, 1230.
© Henry S. Rzepa, 1978-2014. Hide|show Toolbar.