Molecular Modelling Problem.
Electrophilic substitution in furan can be modelled by
protonation. At least three possible protonated isomers are possible; two are shown
below (what is the third?)
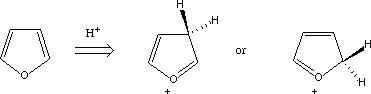
- Calculate the energies of the three protonated forms
of furan at various levels of theory, including MM2 Mechanics
(ChemBio3D), Semi-Empirical (PM6), RHF Ab initio
and correlated B3LYP density functional. Which technique(s) agree with
the known electrophilic substitution pattern of furans in the
2-position. How "expensive" in terms of computer time are the
various techniques?
- What happens with other heterocycles, i.e. pyrroles (N),
thiophenes (S), oxazoles (NO)?
- Does benzo substition on the heterocycle (i.e. benzofuran or indole) change the
pattern?
- Do you get the same prediction if you inspect merely the HOMO (highest occupied molecular orbital) of the reagent?
- If you have performed a Gaussian calculation, you will have the .fchk file. From this you can generate a molecular
electrostatic potential, using the DOS command cubegen 0 potential=scf indole.fchk test.cube. The cube file can then ve viewed
using ChemBio3D or Gaussview. Does the MEP give the same result as the other techniques?
- If you apply a transition state model by using a reaction
coordinate and an electrophile such as nitronium cation, do
the results change?
Instructions and Manuals for using Programs
The problems can be attempted using Gaussian 03
(Ab initio) and the Chemistry SCAN resource. Details on
how to prepare a program input are found here.
(c) Copyright H. S. Rzepa, 1999-2009.