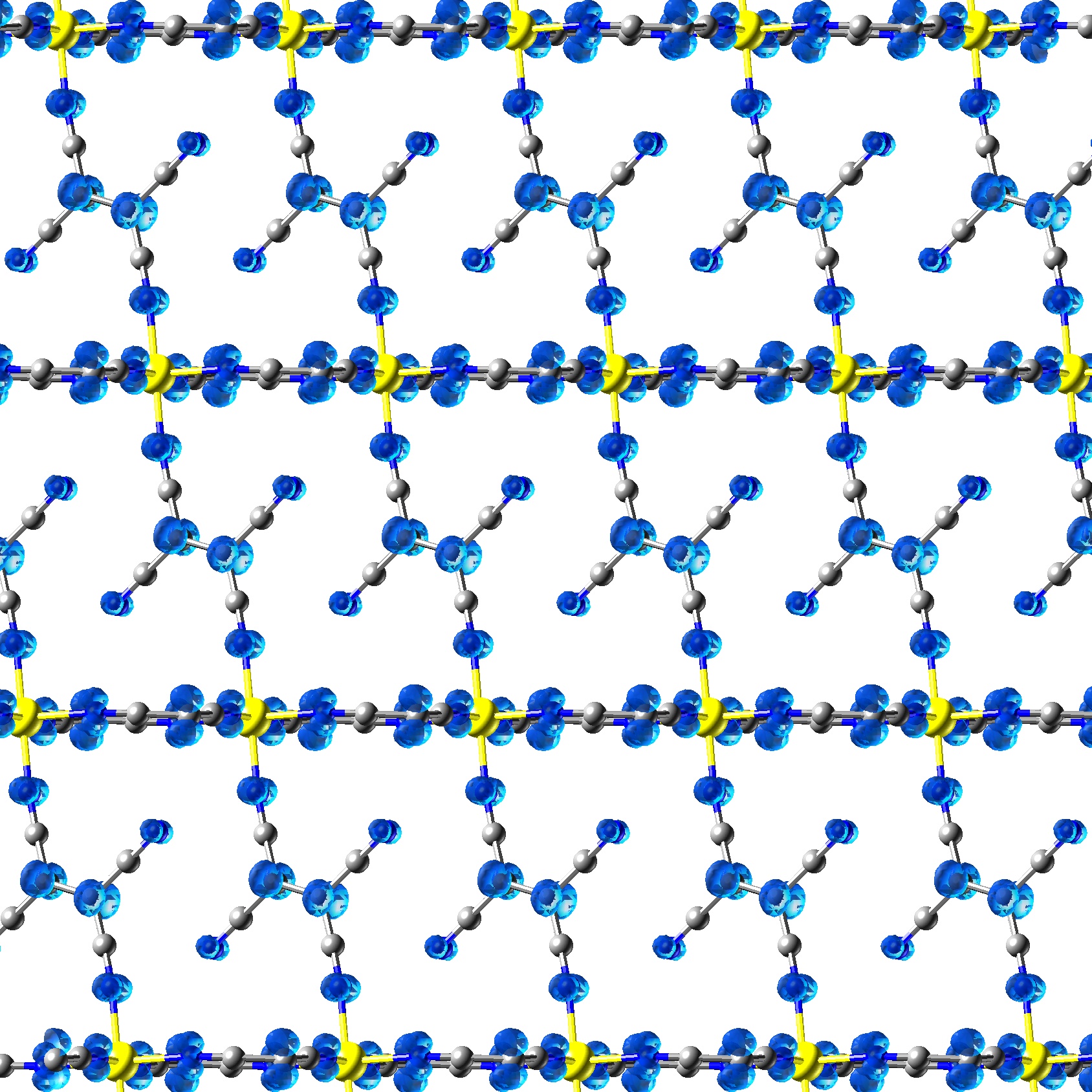
A Previously Proposed Magnetic Structure of Carbon is Unstable
The structure and stability of a pure carbon structure involving mixed sp2-sp3 bonding, long thought to be ferromagnetic, is examined. The structure is found to be unstable with respect to a barrier free transformation to a non magnetic polymorph. Stability of the Ferromagnetic State in a
Mixed sp2-sp3 Carbon System
L. Pisani, Barbara Montanari, Nicholas M. Harrison
Phys. Rev. B (in press 2009)
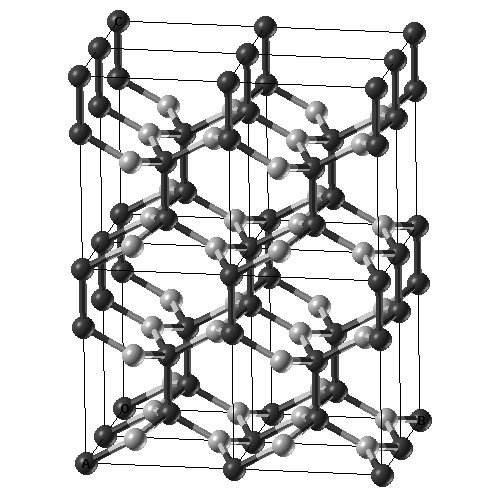
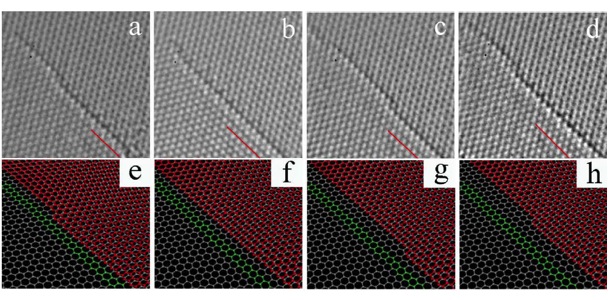
Structural Transformations in Graphene
A combination of hybrid exchange density functional theory and transmission electron microscopy with high spatial and temporal resolution is used to develop a model of the structural transformations in graphene sheets induced by beam damage at 80keV. Structural Transformations in Graphene Studied with High Spatial and Fast Temporal Resolution
Jamie H. Warner, Mark H. Rummeli, Ling Ge, Thomas Gemming, Barbara
Montanari, Nicholas M. Harrison, Bernd Buchner, G. Andrew D. Briggs
Nature Nanotechnology (in press 2009)
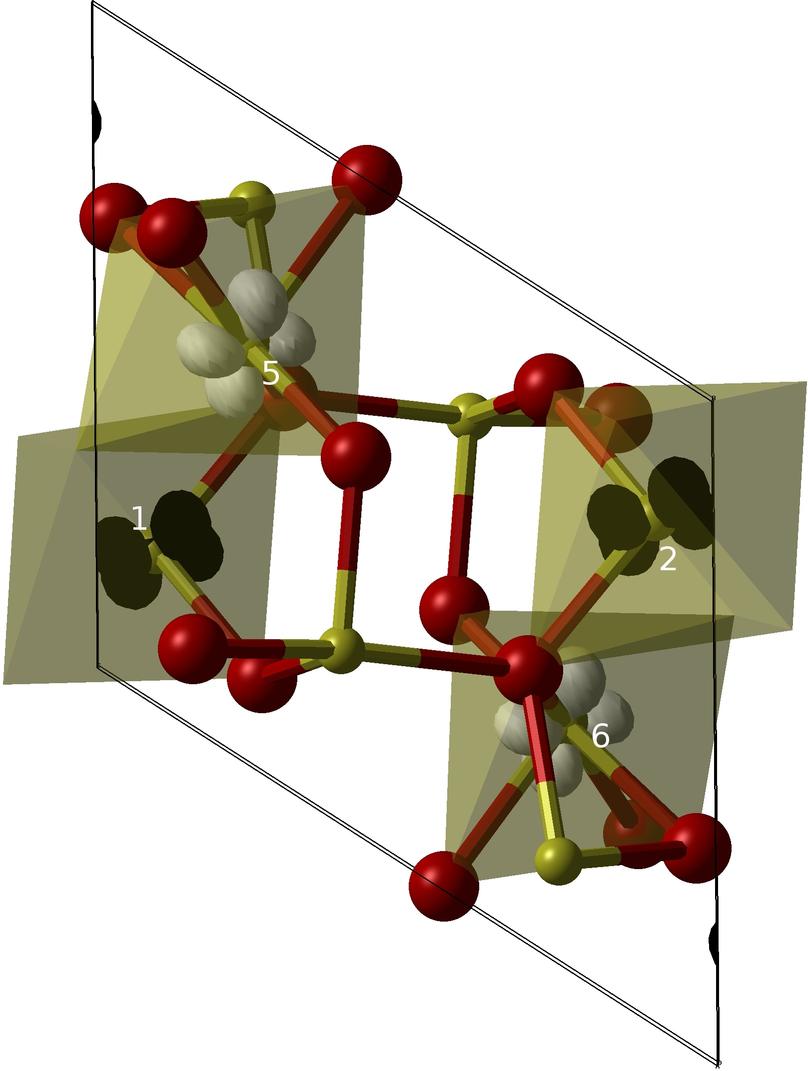
Bipolarons, Orbital Ordering and Magnetism in Ti4O7
Hybrid exchange density functional theory calculations are used to provide a consistent description of the low, intermediate and high temperature phases of Ti4O7. Strong on-site Coulomb interactions and electron phonon coupling produce zero spin polarons in the LT phase. In the IT phase a subset of the bipolarons dissociate but the electrons remain highly localised. The Metal-Insulator Transition in the Ti4O7 Magneli Phase
Leandro Liborio, Giuseppe Mallia, Nicholas M Harrison Phys. Rev. B 79 245133 (2009)
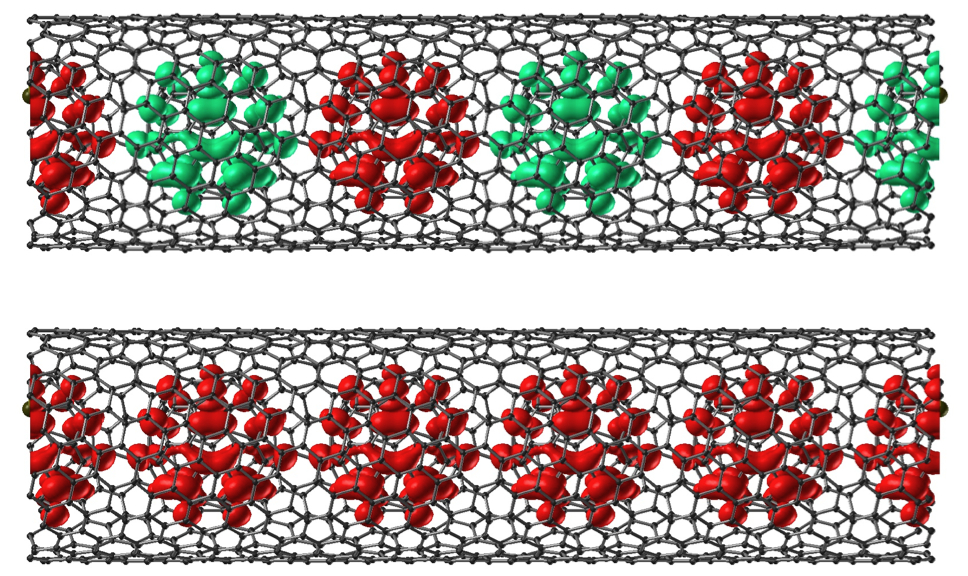
Spin Transport in Carbon Peapod Structures
Electron spins on fullerene cages may be a suitable system for realising a scalable quantum bit (qubit) array. A possible implementation of this concept is to use carbon nanotubes to encapsulate a chain of endohedral fullerenes such as Sc@C82 to form a peapod structure. The spins generated by electron donation from the Sc ion are localised on the fullerene chains. The spin interactions, electronic structure and dynamics of this system has been studied.
Effects of Doping on Electronic Structure and Correlations in Carbon Peapods
L. Ge, John H. Jefferson, B. Montanari, N. M. Harrison, D. G. Pettifor and
G. A. D. Briggs, ACS Nano ASAP (2009)
DOI 10.1021/nn8008454
Modelling Spin Interactions in Carbon Peapods using Hybrid Exchange Density Functional Theory
L. Ge, B. Montanari, J. H. Jefferson, D. G. Pettifor, N. M. Harrison,
G.A.D. Briggs, Phys. Rev. B 77 235416 (2008)
Dynamics of Paramagnetic Metallofullerenes in Carbon Nanotube Peapods
J. H. Warner, A. A. R. Watt, L. Ge, K. Porfyrakis, T. Akachi, H. Okimoto, Y. Ito, A. Ardavan, B. Montanari, J. H. Jefferson, N. M. Harrison, H. Shinohara, G. A. D. Briggs Nano Lett. 8 1005 (2008)
Molecular Magnets
Materials based on transition-metal-tetracyanoethylene (TCNE) are of particular interest as molecular magnets as they display a rich range of magnetic behaviour and have relatively high critical temperatures. The vanadium material is remarkable in that it has magnetic order above room temperature but is X-ray amorphous. Hybrid exchange DFT has been used to develop a structural model and to explain the exceptionally strong magnetic coupling. Density functional study of the magnetic coupling in V(TCNE)2
Giulia C. De Fusco, Leonardo Pisani, Barbara Montanari, Nicholas M. Harrison , Phys. Rev. B. 79 085201 (2009)