In this exercise you will calculate the wavefunction of an MgO crystal by loading a CRYSTAL input file: MgO.inp
Starting DLVisualize
Run a CRYSTAL calculation:
Calculate->CRYSTAL->Run SCF Input file
Click on Browse
Select MgO.inp
Select OK
Select OK
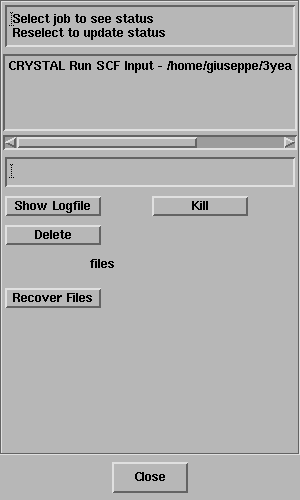
The Job List panel will open automatically and look something like this;
Select the job and the status line should
report "Job has completed" - like this;
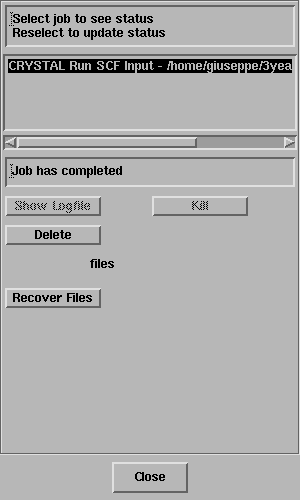
Click on Recover Files.
A model of MgO should have appeared in the DLV 3DView window.
This structure should look reasonably familiar it is the rocksalt
lattice - with oxygen ions (red) and magnesium ions (green) drawn at their
ionic radii.
On main panel the square icons are toggles which control how the 3D structure is displayed. A help message is displayed when you pass the mouse over each button. At first you are in rotate mode and can spin the structure by dragging with the right mouse button. Try changing to scale mode and translate mode etc.
Open the Structure Display panel
Display->Structure
Try out the controls on this panel - you can change the size of the atoms, display bonds, multiple unit cells etc etc.
Turn on label lattice to see the a, b and c lattice vectors labels.
The same lattice may also be described:
Use the na, nb and nc sliders on the Display->Structure panel to draw multiple cells.
Give the current model a suitable name. This will be used as the
label for this model in subsequent CRYSTAL simulations.
Edit -> Model -> Name
Change "Model_1" to something suitable; say "MgO".
Note:
For the change to take effect you must press Return.
You'll see the name of the graphics window change to "DLV 3DView - MgO";
A second window will open displaying the output from the CRYSTAL calculation (the LogFile).
It should look something like MgO.txt
Take some time to read through the LogFile.
Which is the hamiltonian/method adopted in the calculation?