In this exercise you will perform calculations of properties by exploiting the wavefunction of an MgO crystal obtained in the previous exercise.
A prerequisite for calculating properties is to analyse a wavefunction,
otherwise the properties items are grayed out.
Calculate->CRYSTAL->Analyse Current Wavefunction
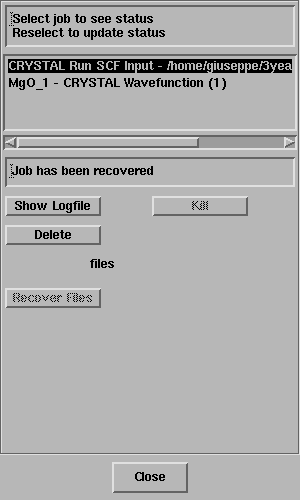
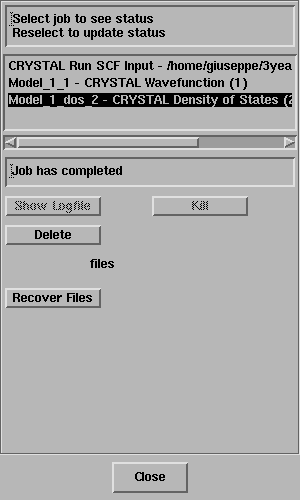
The Job List panel will open automatically and look something like this;
Select the job and the status line should
report "Job has completed" - like this;
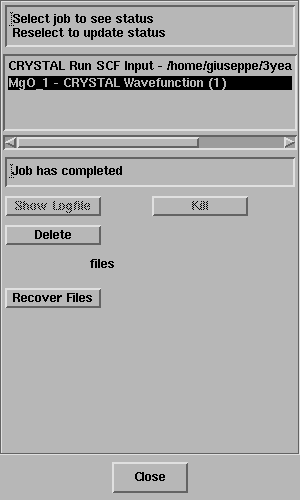
Click on Recover Files.
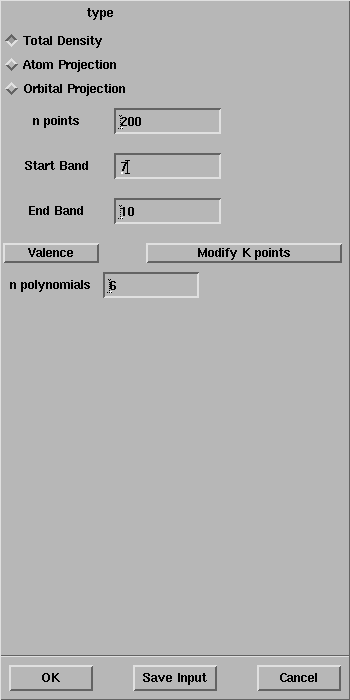
Select Calculate -> CRYSTAL -> Properties -> Density of States.
The CRYSTAL Density of State panel will open:
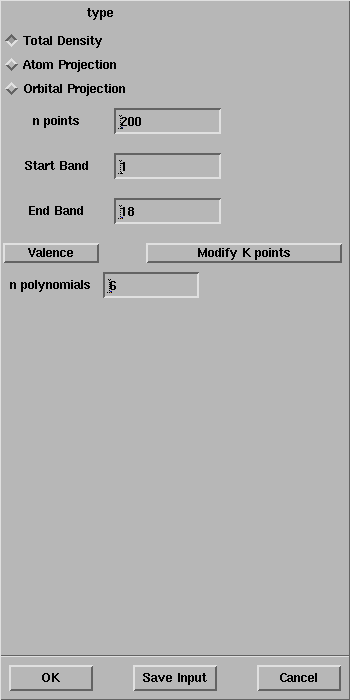
Select OK and the properties calculation will start as evident from the Job List panel
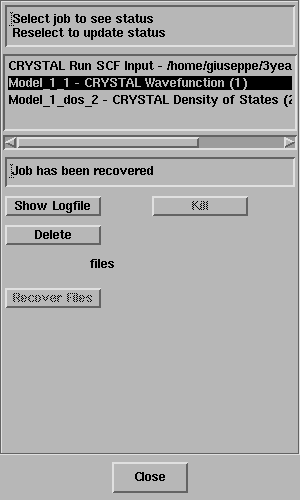

Exercise 2: Repeat the previous exercise,
and press Valence in the CRYSTAL Density of State panel.
Does the Start Band change with respect to the previous exercise?
Does the End Band change too?
Why? How many electrons are there in a MgO primitive cell?
How many valence electrons? How many core electrons? How many are the bands
occupied by the core electrons?
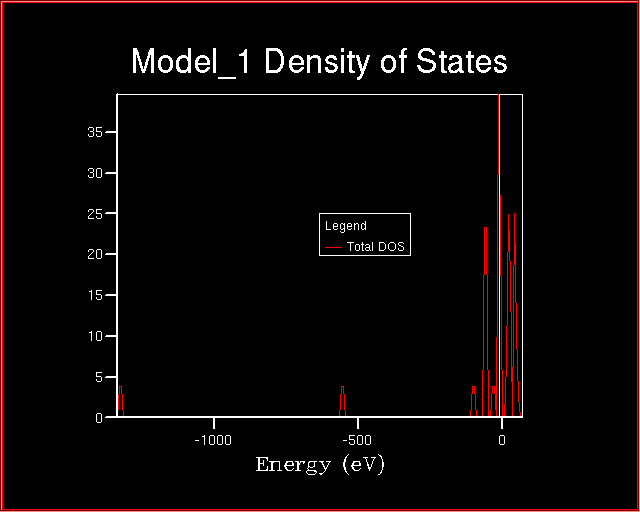
At the end of the calculation, the Density of States looks like:

When comparing the two plot of the Densities of States, many changes are
evident. The most significant is related to the energy range.
What conclusions can be drawn about the valence and the core levels?
Exercise 3: Repeat the exercise 1, select the Atom Projection
toggle and follow the instructions in the window.
Ensure that the Start Band and the End Band are equal to 1 and 18,
respectively.
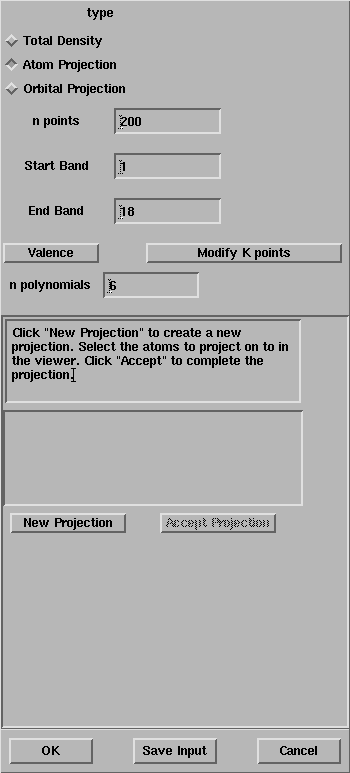
After the selection of the magnesium and the oxygen atoms, the window
looks like:
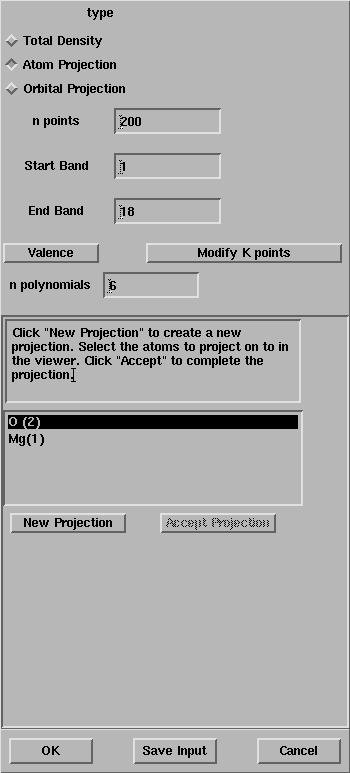
Press OK and the properties calculation will start.
When the job has completed, click on Recover Files
and in the 1D Data Display panel a
new data set becomes available.
Select the new set and Draw 1D data.
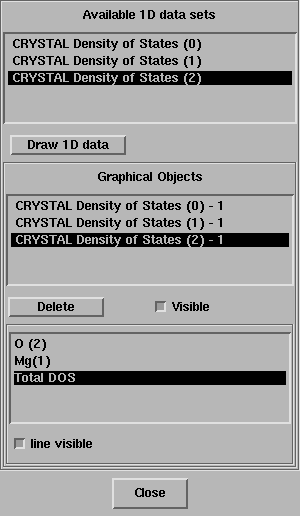
Select TOTAL DOS in the bottom box of the 1D Data Display panel turn off the option line visible.
The plot becomes as follows:
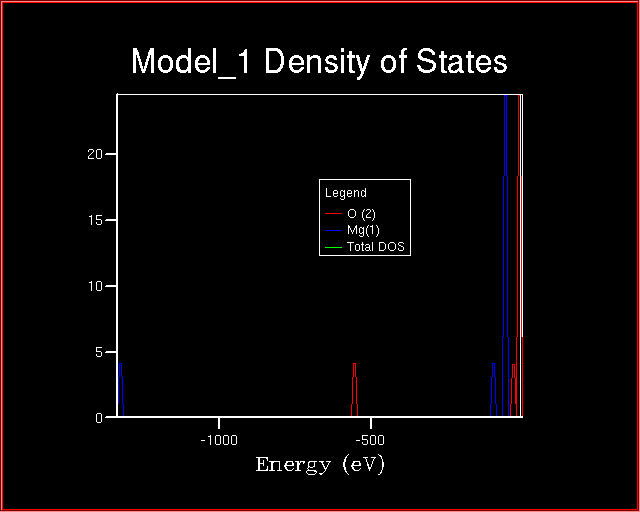
Which is the relation between the TOTAL DOS and the two ATOM projections of the TOTAL DOS?
Why the core states of the Magnesium ion are lower in energy
than the Oxygen ones?
Exercise 4: Repeat the exercise 3,
and press Valence in the CRYSTAL Density of State panel.
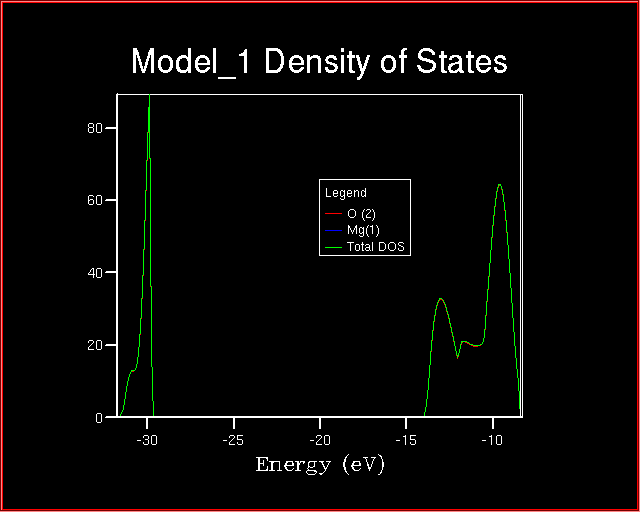
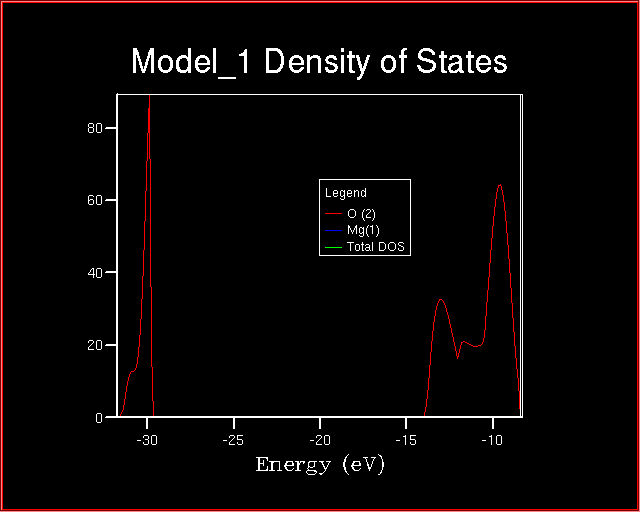
When comparing the Total Density of State and the Projected one,
is it correct to state that the two valence bands
has magnesium character and are derived from magnesium orbitals?
If not, explain why?
Exercise 5:
Repeat the exercise 1
and press Valence in the CRYSTAL Density of State panel.
Select the Orbital Projection
toggle and follow the instructions in the window:
After the selection of the oxygen atoms, the window looks like:
The list of all the atomic orbitals of the oxygen atom is given in the box,
select all the s functions.
Press OK and the properties calculation will start.
When the job has completed, click on Recover Files
and in the 1D Data Display panel a
new data set becomes available.
Select the new set and Draw 1D data.
The following plot appears:
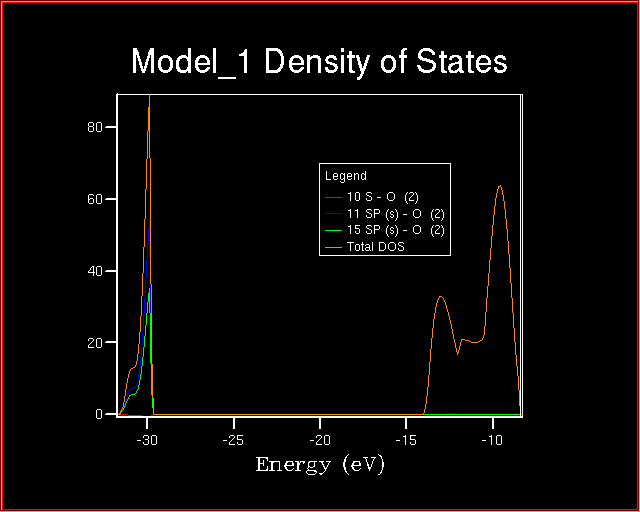
The lower valence band is derived from Oxygen-s orbitals.
As evident from the plot, the shape of the total density of states
can be obtained when the Oxygen-s orbital projections are added up.
Why is the red line projection irrelevant?
Exercise 6:
Repeat the exercise 1
and press Valence in the CRYSTAL Density of State panel.
Select the Orbital Projection
toggle and follow the instructions in the window:
After the selection of the oxygen atoms, the window looks like:
The list of all the atomic orbitals of the oxygen atom is given in the box,
select all the functions different from s.
Press OK and the properties calculation will start.
When the job has completed, click on Recover Files
and in the 1D Data Display panel a
new data set becomes available.
Select the new set and Draw 1D data.
Is the upper valence band derived from Oxygen-p orbitals? Can the shape of the total density of states be obtained when the Oxygen-p orbital projections are added up?
Exercise 7: Calculate the band gap of MgO?
In insulators and semiconductors, the band gap is the energy difference
between the top of the valence band and the bottom of the conduction band.
Select Calculate -> CRYSTAL -> Properties -> Density of States.
In the CRYSTAL Density of State panel,
set the Start Band and the End Band equal to 8 and 11,
respectively.
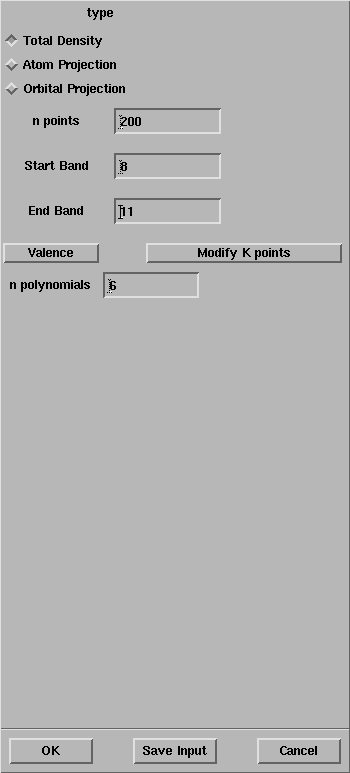
Why the End Band must be equal to 11 in order to compute the bandgap?
Is the 11th band occupied or virtual?
At the end of the calculation, the density of states looks like:
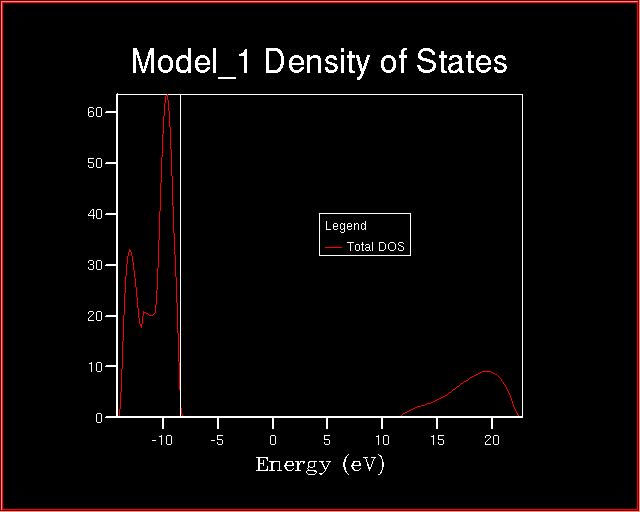
Compare the calculated band gap with the experimental value.