Online Validation and Comparison of Molfile and CML
Molecular Atom-Connection Descriptors
Georgios V. Gkoutos and Henry S. Rzepa
Department of Chemistry, Imperial College London, SW7
2AY.
Peter Murray-Rust
Unilever Centre for Molecular Informatics, Department of
Chemistry, University of Cambridge, Lensfield Road, Cambridge.
CB2
Abstract:
We describe an online resource in the form of this journal
article for comparing the molecular constitution of molecule
descriptors expressed as either the MDL Molfile or CML
(Chemical Markup Language, an XML language). The resource
comprises a set of server-based tools for validating molecule
descriptors expressed in either CML or MDL Molfile syntax, and
for comparing the constitutional identity of any two molecules
so described. We discuss the issues of how XML Schema-based
validation compares with non-XML-based formats such as the
molecular Molfile. This system serves to exemplify a component
of a concept we call the Chemical Semantic Web, in which
journal articles such as this one can be used as a (re)usable
and integrated molecular resource.
Keywords:
SMILES, Comparison of MDL Molfile, CML, Molecular resources,
Semantic Web, XSLT, XSD.
Introduction
The Semantic Web1 represents a
coherent and systematic attempt to create a set of technologies
and standard protocols for expressing information on a global
scale, and in a machine understandable and processable form.
Such information and the resources to process it could take
many forms, including for example journal articles such as this
one. Any implementation of a chemical semantic Web in
particular must also include procedures for the recognition of
valid chemical information components or "objects" and provide
recognisable methods for processing such objects and
establishing their relationship to the global body of chemical
information and resources. One of the best defined object
models in ontological terms is the "molecule", of which at
least 30 million have been formally described in some manner.
Molecular descriptors can take many (but often incompatible)
forms one of which is a list of atom-coordinates and
bond-connections, expressed in a variety of syntactic forms.
Various methods for harvesting and aggregating molecular
information in this form have been implemented over the years,
originally taking the form of human abstraction of information
found in journals, which includes crystallographic,
spectroscopic and other molecule property data. With the advent
of the Internet, various attempts to increase the automation of
this by indexing or abstracting Web-based documents and
resources have been made.2
We have previously reported one such robot-based system
called ChemDig3, which acts as a
molecular harvester and can output the results of this process
into various molecular databases for deposit. This agent was
designed to recognise molecular information by its association
with very simple metadata, the Chemical MIME types.4 These types include various older
formats such as the MDL Molfile5.
This harvesting procedure did implement some simple checks that
the syntax of the file harvested represented a recognisable
Molfile (necessary because human authoring of this format will
often result in minor syntactic variation) but it did not
per se check that the atom/bond list represented any
form of "valid" molecule as defined by a specified scheme (such
as one for example expressing the rules of valency). No
effective rules can be easily imposed at the (human or software
based) authoring point of creating a list of atoms/bonds, such
as the order in which they are specified. Because of this it
was not possible in general to establish what relationship any
one molecule might have to previously harvested molecules.
There was clearly a need to establish whether any two molecules
were constitutionally identical (duplicates), whilst
recognising the ultimate need to be able to compare more subtle
features such as three dimensional stereochemistry, tautomerism
and other aspects of chemical perception. Although commercial
software such as the Daylight toolkit can internally normalise
some types of molecular representations to achieve both these
tasks, these tools are not freely available as an Internet
resource, and they are not readily extensible to inclusion of
other syntactic representations.
This article constitutes both an on-line resource for and a
description of how the constitution of two molecule objects
defined using either MDL molfile CML syntax (the latter based
on XML or eXtensible markup language)6,7 can be
compared. We have used generic OpenSource XML software and
procedures wherever possible to ensure that these tools are
modular, extensible, and compatible with other XML
resources.
Molecular Validation
In general, the process of ensuring that a file or document
containing a molecular descriptor is valid has three parts.
- Firstly, the syntax has to be identified (achieved on the
Web using MIME headers4) and
that if possible the file is complete with an appropriate
header and terminator. Thus a molecule descriptor in a MDL
Molfile should include a termination with M END; in
CML there should be a closed container of the type
<molecule ...></molecule>
- The remaining structure of the file should also follow
the expected syntactic rules. Thus a Molfile is formally
defined syntactically5, although
in practice much minor variation is found, and accordingly
most checks take a more relaxed form. Essentials such as the
presence of precisely one line of text per declared atom can
be checked (and is actually non trivial given that any of
three combinations of character defining the end of a line
might be present) but tests for e.g. the presence of allowed
values for any (2D or 3D) coordinates may be often omitted.
This can lead to substantial ambiguity; does the presence of
the value 0.00000 for all the atomic third coordinates mean
this value is unknown or that the molecule is planar? It is
probably true that few software solutions for reading (and
writing) a Molfile adhere strictly to the published
specifications5, and that any
stricter validation will normally be assumed to be required
in any subsequent processing of the information (such as
deposition into a repository). The use of an XML language
such as CML for this type of validation has one distinct
advantage. The formal specification7 is available in a form (the XML Schema
definition) which can be processed using standard software
tools and which therefore will not need any custom software
written. This enables much stricter validation to be
routinely attempted; for example there is no ambiguity about
whether a given coordinate is part of a 2D definition (the
third coordinate is thus unambiguously not present) or a 3D
set (in which case a value of 0.00000 means precisely that,
and not that it is unknown). Use of schema validation ensures
that all mandatory data is present, that attributes of any
data container (element) are present if required, and most
importantly that the data when present has the correct
datatype. An example of the latter would be that an atom list
contains only types selected from an allowed enumerated list
(such as the periodic table). In this implementation, we
invoke a loose validation for the MDL molfile (checking
essentially for no truncation and the presence of correct
syntax for the atom and bond descriptor lines, but not
necessarily correct values such as bond and atom types) and a
precise validation of CML files according to the published
schema.7
- The third level of validation is related to chemical
perception. Simple examples might be to perceive what the
implied valence at any atom might be in relation to any
declared (or undeclared) hydrogen atoms present, and to flag
if a valence exception may have occurred. More complex
examples might be "impossible geometries" (for example the
approach of two atoms to an unrealistic distance), or even
higher levels of perception relating to the identity of two
different tautomeric representations of what in effect is the
same molecule. We have not attempted this level of validation
in the present tool, although we recognise its importance and
will report in the future on implementations using XSLT-based
validation.
Unique Molecular Descriptors
Two basic types of unique molecular descriptor have been
developed in the past. The first, typified by the CAS registry
number, is based on a dictionary lookup of a text-based
identifier which in itself carries no semantic information. By
2002, some 30 million molecules had been allocated such a
registry number. The second type is based on creating a unique
descriptor using a defined algorithm which generates a
semantically meaningful string. In theory, anyone with access
to the definition of the algorithm should be able to generate
the same identifier. The best known such implementation is
SMILES8 (Simplified
Molecular Input Line Entry
Specification), which can itself take both un-normalised
(non-unique) and canonicalised (unique) forms. The former,
although carrying semantic information about the atoms and
bonds in a molecule (but not their three dimensional
relationships) will depend on the (non-unique) order in which
the atoms are specified. The process of canonicalisation
ensures that the order and description of these properties is
transformed to a unique form. Unfortunately, the published
SMILES algorithm for generating this unique form has over the
years diverged slightly from the current implementation in the
Daylight toolkit. Other implementations of the published SMILES
algorithm such as that in the JME tool written by Ertl8 also diverge and an open definition of
the SMILES implementation in JME is not available. The
consequence is that several possible "unique" SMILES strings
can be generated for any given molecule. What remains true
however is that if canonicalised SMILES strings for any two or
more molecules are generated using the same tool, then
comparison of these strings should provide a clear indication
of whether or not the molecular constitutions (a list of
non-hydrogen atoms and their bond connectivities) of two or
more molecules are identical. Advanced features such as
perception of stereochemistry, tautomerism, aromaticity and
other chemical features, or normalisation of hydrogen counts
etc, are currently less well handled and are not implemented
here, as noted above in regard to validation. Possible
solutions to this last issue are however noted in the
discussion below.
Procedures
We chose to implement procedures for two types of molecular
connection table formats, the MDL molfile,5 and one XML implementation, the CML
format.6,7
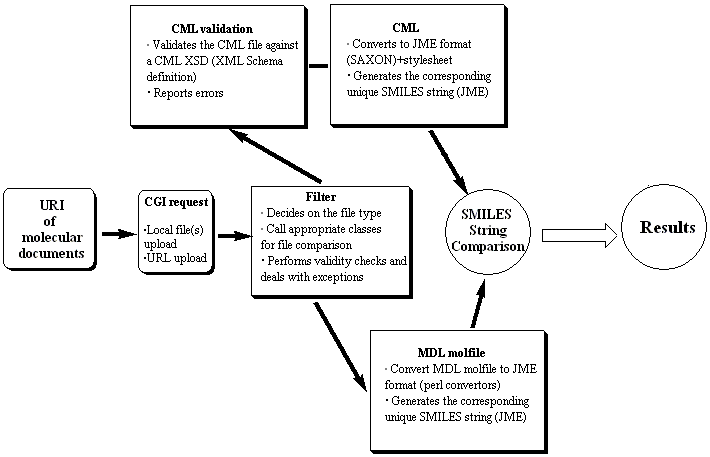
Scheme 1. A schematic presentation of the online
transformation and comparison tools
The procedures outlined in scheme 1 are elaborated
below;
- An interactive form to invoke a CGI program resident on a
server with an option of selecting a target file either
locally or from a URI address. The file is retrieved and
saved locally on the server in a temporary directory.
- For a CML 1.0 file, an option includes formal validation
against a CML Schema7 to ensure
both XML well-formedness for the file and a limited degree of
chemical validation (such as ensuring that the data type for
the identity of each defined atom conforms to an enumerated
list of the chemical elements and specified additional atom
types). This task makes use of the opensource XML parser
Xerces,10 which accepts an XML
data file as input and validates it against a specified XSD
schema file.7 The specific
syntax for this step is shown below;
dom.ASBuilder -F -a schema.xsd -i
document_to_be_validated.xml
Both Xerces and the Class dom.ASBuilder
interface to it are downloadable.10 In principle, other XML Schema
validators could be used if required, and appropriate XML
schemas could be implemented for other XML lanaguages.
-
Options for generating a canonical SMILES string from one
local MDL molfile or CML file or from a URI address
specifying the location of this file, or acquired by
uploading a local disk-resident file. This task makes use
of two transformation tools.
- A Perl script is used to convert a Molfile to a JME
string, which will be passed to the JME parser Java class
for conversion to the JME form of a canonical SMILES
descriptor. Alternatives to this particular procedure
could be to use the SMILES processor present in the
opensource CDK (Chemical Development toolkit)11, available as Java classes, or
the OpenBabel toolkit12.
- SAXON13 is a collection
of tools for processing XML documents, including an XSLT
processor for transforming XML data. This was used in
conjunction a XSLT stylesheet written for the task of
converting a CML 1.0 file6
to JME form. An alternative, not available when this
project was commenced, would be to use the CDK toolkit
for this conversion11. A
Java Class Convert2Mol.class was written to call
the com.icl.saxon.StyleSheet Saxon class, which
transforms a CML 1.0 file using the stylesheet cml2jme.xslt.
Extension of this stylesheet approach to other
post-processing transforms can be envisaged, for example
to define the SVG graphical elements corresponding to a
2D representation of the molecule.14
- The user has also the option of comparing two separate
Molfile or CML files. Two file locations are provided by the
user (both local or both specified via a URI) and after the
conversion processes for both files are completed, the
corresponding canonical SMILES string for each is generated,
compared and the result presented to the user. The CML
validation option is particularly useful for checking older
CML (such as produced by e.g. JChemValidate15 using Perl script converters) since
the newer Schema approach7
ensures rigorous conformance for the generic XML syntax and
form (ensuring for example that only controlled values of the
XML-attributes appropriate for the CML definition are present
and have the correct data types). These checks can also be
used to verify integrity of ftp transfers and occurence of
duplicate entries on the Web. Such operations, although
trivial for small molecules, still require human intervention
via visual inspection and hence are prone to error. Larger
molecules are of course more difficult to verify in this
visual sense.
Table 1 presents a summary of the classes involved in these
tools.
| Table 1. SmilesConvert and SmilesCompare
classes |
| SmilesConvert and SmilesCompare classes |
Description |
| Molfile2SmilesConverter.class |
Retrieves and decodes the URI or local MDL molfile,
ensure its validity and initiates the conversion and
comparison procedures. |
| CML2SmilesConverter.class |
Retrieves and decodes the URI or local CML file, ensure
its validity and initiates the conversion procedures. |
| mol2jme.pl |
A perl script that converts the MDL molfile to the JME
format |
| CML2JME.class |
Converts a CML 1.06 file
to a JME format using SAXON XSLT processor |
| JME.class |
Generates a canonical SMILES string of the
corresponding structures |
| Convert2Smiles.class |
Calls appropriate classes for conversion and generation
of canonical SMILES string |
| Runcomparison.class |
Compares two canonical SMILES strings and presents the
results to the user |
Form 1 and Form 2
are included as a integral part of this article as working
examples and define two interactive areas that invoke our
procedures for processing of respectively a single file and
comparison of two separate files using JME-based
canonicalisation of the two structures. Form 1 can be used to
also validate a CML file against the current (CMLCore) schema
definition for the latest specification of CML.7
| Form 1.
Validate (optionally) and Generate Canonical Descriptor
from CML/MDL molfile |
|
|
| Form
2. Comparison of two CML/Molfiles. |
|
|
Discussion
The procedure described here is just one of several
approaches to the problem of identifying how two molecular
descriptors may relate to each other; are they different
constitutional molecules or merely different syntactical
representations of the same constitutional molecule? The use of
the generic name of a chemical structure is one of the most
common ways of attempting to exchange such molecular
information. However, generic names can have many variations
corresponding to a single chemical structure. Many chemical
structures, especially when novel and unpublished, may not have
a generic or chemical name. Ideally, systematic naming rules
(such as Autonom16) would result
in a unique single name which can also be regarded as a unique
descriptor, just like the molecular structure itself. There is
no guarantee that any such name would be incorporated into a
descriptor such as the MDL molfile, or if it were, to actually
correspond to the subsequent definition of atoms and bonds.
Furthermore, the proprietary nature of such software often
results in incompletely documented algorithms, and divergences
of the type already noted for SMILES descriptors. Hence, there
is a clear need for a unique and public domain identifier for
chemical substances that can be used in printed and in the
present context, electronic data sources. A particularly
promising and potentially more general solution which is under
development is IChI (IUPAC Chemical Identifier), a
non-proprietary chemical identifier that would be easy to
generate, expressive and unambiguous17. Though not available for public use
when this project was undertaken, we anticipate that it has the
potential to become an essential and importantly, an openly
documented alternative to the SMILES approach taken here.
Additionally, IChI will have significant chemical perception
capabilities, including e.g. detection of aromaticity,
tautomerism, and other structural features. Because the CML
format and IChI are both XML-compliant, it is trivial to
incorporate the latter within the former; an extension not
easily possible in the more proprietary and older formats such
as MDL molfile V2. Currently, the IChI algorithm is still being
tested and finalised and for this reason, the procedures
described here currently rely on SMILES generation for
providing a procedure for establishing the constitutional
identity or non-identity of two separate molecule descriptor
files. Nevertheless, the IChI method could easily be
incorporated as an alternative to SMILES once it is publicly
available. Once a robust mechanism is in place for determining
molecular uniqueness, procedures such as merging information
from multiple sources becomes possible. Information relating to
the same molecule can be identified and supersets created; XML
technology appears to be the appropriate method for capturing,
merging and if necessary filtering/transforming the information
into new forms.
Conclusions
We have described a molecular resource for partial validation
of a molecular atom-connection descriptor and for comparison of
two such descriptors which may have been sourced from
unconnected locations on the Web. The resource takes the form
of this journal article and has been constructed using standard
tools and protocols. Such integration between the primary
scientific literature and semantically rich resources are a key
feature of the Semantic Web. As such resources become more
readily available resulting from nodes in scientific grids
which are increasingly being funded, so methods for their
discovery need to be developed. This will involve registration
procedures and metadata based descriptions of their properties
invoked by software agents. Standards such as WSDL (Web
services discovery language) and OGSA (Open Grid Services
Architecture)18 will play an
important role in creating a framework for discovery and
invocation of molecular resources such as those described here.
A pilot project describing the use of such protocols in
creating a Chemistry Web services node is already under
way.19
Acknowledgements
One of us (GVG) thanks Merck Sharp and Dohme and the EPSRC for
the award of a studentship.
References and Notes
- T. Berners-Lee and M. Fischetti, 1999, "Weaving the Web:
The Original Design and the Ultimate Destiny of the
World-Wide Web", London: Orion Business Books.
- For reviews of various approaches to this, see J.
Gasteiger and T. Engel (Eds), "Chemoinformatics - From Data
to Knowledge", Vols 1 and 2, 2003, (Wiley), in press.
- G. V. Gkoutos, C. Leach and H. S. Rzepa, New. J.
Chem., 2002, 656-666.
- H. S. Rzepa, P. Murray-Rust and B. J. Whitaker, J.
Chem. Inf. Comp. Sci., 1998, 38, 976-982.
- A. Dalby, J. G. Nourse, W. D. Hounshell, A. Gushurst, D.
I. Grier, B. A. Leland and J. Laufer, J. Chem. Inf. Comp.
Sci., 1992, 32, 244-255.
- P. Murray-Rust and H. S. Rzepa, J. Chem. Inf. Comp.
Sci., 1999, 39, 928
- P. Murray-Rust and H. S. Rzepa, J. Chem. Inf. Comp.
Sci., 2003, 43, in press. The CMLCore XSD Schema
file is available at http://www.xml-cml.org/
and http://cml.sourceforge.net/
- D. Weininger, A. Weininger and J. L. Weininger, J.
Chem. Inf. Comp. Sci., 1989, 29, 97-101;
ibid, 1988, 28, 31-36.
- P. Ertl and O. Jacob, Theochem, 1997, 419,
113-120.
- Xerces 2.3; http://xml.apache.org/xerces2-j/
- C. Steinbeck,J. Chem. Inf. Comp. Sci., 2003,
43, in press; C. Steinbeck and E. L. Willighagen
(Eds), The Chemical Development Kit, http://cdk.sourceforge.net/
- OpenBabel Project: http://openbabel.sourceforge.net/
- G. V. Gkoutos, P. Murray-Rust, H. S. Rzepa, C. Viravaidya
and M. Wright, Internet J. Chemistry, 2001, article
12. See also http://www.ch.ic.ac.uk/rzepa/xml/
- M. H. Kay, http://saxon.sourceforge.net/
- G. V. Gkoutos, P. Kenway, P. Murray-Rust, H. S. Rzepa and
M. Wright, Internet J. Chem., 2001, 4, article 5.
- J. L. Wisniewski, Abstr. Pap Am. Chem. Soc., 2001,
222: 2-Cinf Part 1 Aug 2001. see http://www.beilstein.com/products/autonom/
- S. E. Stein, S. A. Heller and D. V. Tchekhovskoi, Abs.
Pap. Am. Chem. Soc., 2001, 222, Chicago, IL,
United States, August 26-30, CINF-005. See
http://www.iupac.org/projects/2000/2000-025-1-800.html
for details of the IChI Project.
- D. Talia, IEEE Internet Computing, 2002, 6,
67-71. For details of Open Grid Services Architecture, see http://www.globus.org/ogsa/
- P. Murray-Rust, M. Osmond, H. S. Rzepa and M. Wright, the
Hydra Project: http://hydra.ch.ic.ac.uk/