Unlike thiane oxide and other dithiane monosulphoxide systems, 1,3-dithiane monosulphoxide exhibits a small preference in solution (84:16 at -80 deg.C) for a chair conformation with equatorial sulphoxide (DG0 = 0.64 kcal mol-1 at -80 deg.C; DG = 11 kcal mol -1).6
Monooxidation of 2-substituted 1,3-dithianes gives predominantly the trans stereochemical arrangement in the products for simple alkyl or aryl 2-substituents,2,3 suggesting equatorial oxidation of a chair conformation of the substrate in which the 2-substituent is also equatorial, presumably the preferred conformation, or axial oxidation of a chair conformation of the substrate in which the 2-substituent is also axial. We have shown that this pattern of reactivity holds for 2-alkyl-1,3-dithianes and for 2-acyl-2-alkyl-1,3-dithianes.2,3,5
It is particularly interesting that a second oxidation of 1,3-dithiane monosulphoxide, which occurs at the unoxidized sulphur atom, takes place under most conditions to give predominantly the trans disulphoxide, in which, in a chair conformation, one sulphoxide is equatorial and the other axial.2,5,7 Molecular mechanics calculations have suggested that the trans disulphoxides of dithioacetals are more thermodynamically stable than the cis (DG0 = 0.80 kcal mol-1).8 We have shown that this pattern of reactivity also holds for 2-alkyl-1,3-dithianes and for 2-acyl-2-alkyl-1,3-dithianes.5 This second oxidation could occur by equatorial oxidation of the substrate conformation containing axial sulphoxide, the less stable arrangement, or by axial oxidation of the more stable equatorial sulphoxide conformation, or both.
In order to investigate further we have prepared a 1,3-dithiane containing a 5-tert-butyl substituent in five steps from diethyl 2-tert-butylmalonate (Scheme 3).9,10 Lithium aluminium hydride reduction to the diol and ditosylation were followed by displacement of both tosylate groups using potassium thioacetate. Lithium aluminium hydride reduction gave the dithiol, which was converted into 5-tert-butyl-1,3-dithiane by platinum catalysed coupling with diiodomethane.11 Examination of the 200 MHz 1H NMR spectrum indicates that this compound exists in solution with the tert-butyl group predominantly in the equatorial position (the signal corresponding to the proton at C5 appears as a triplet of triplets with coupling constants of 2.5 and 11 Hz).12 High temperature NMR studies have also indicated conformational inflexibility in 5-tet-butyl-1,3-dithianes.10
Monooxidation of 5-tert-butyl-1,3-dithiane with sodium periodate in methanol/water solution is facile at room temperature, providing the monosulphoxide in 50% yield after four hours. The 250 MHz 1H NMR spectrum of the monosulphoxide products suggests a diastereoisomeric ratio of ca. 11:1. Single crystal X-ray structure determination of the major isomer shows the sulphoxide oxygen atom and the tert-butyl group to have a cis relationship in a chair conformation with diequatorial substituents, indicating sulphur oxidation from the equatorial direction. Evidence that equatorial monooxidation is general for 1,3-dithiane systems is provided by oxidation of 2-butanoyl-2-tert-butyl-1,3-dithiane, where the major product now has a trans relationship between the equatorial sulphoxide oxygen atom and the equatorial tert-butyl group, in a chair conformation.3
Oxidation of 5-tert-butyl-1,3-dithiane to the disulphoxide with sodium periodate in methanol/water solution proceeded smoothly overnight, giving the disulphoxide product in 49% yield as a >13:1 mixture of diastereoisomers. As expected, single crystal X-ray structure determination of the major isomer shows the compound to have the trans dioxide stereochemistry and an equatorial tert-butyl group. The second oxidation has therefore indeed taken place from the axial direction.
In view of the small expected difference in thermodynamic stability between the equatorial and axial monosulphoxides,6 the high diastereoselectivities observed in the oxidation of 5-tert-butyl-1,3-dithiane are perhaps surprising, given that sulphur oxidation by sodium periodate is thought to occur under product development control.13 Consideration of the anomeric effect leads to the suggestion that the axial lone pair should be the more reactive in both the 1,3-dithiane and 1,3-dithiane 1-oxide systems.7,14 Consideration of molecular orbitals may provide a more satisfying analysis. One would expect some interaction to occur between the lone pair orbitals on the two equivalent sulphur atoms of a 1,3-dithiane. Transannular sigma-interaction between the two equatorial lone pairs and ¼-type interaction between the two axial lone pairs would give rise to new pairs of molecular orbitals. The more effective interaction will lead to the highest energy orbital, and if this is the sigma-interaction between the equatorial lone pairs, then an equatorial lone pair will be the most nucleophilic. In the case of the equatorial monosulphoxide, sigma-interaction between the remaining equatorial lone pair and the sulphoxide bond would be expected to be much weaker. ¼-Type interaction between the axial lone pairs would then cause these to be the more nucleophilic.
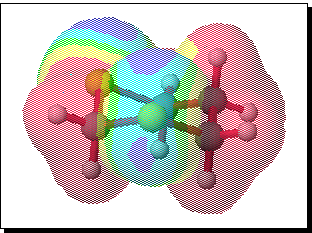
In an attempt to address this aspect, we have performed a series of MOPAC/PM3 and MULLIKEN ab initio calculations to try to identify the origins of the selectivity. In the first instance, ab initio calculations at the 6-31G* level were performed using the CACHe Mulliken system, with full minimisation of the geometries of 1,3-dithiane and its mono-oxide. The analytically calculated molecular electrostatic potentials for both species were evaluated and plotted as a colour coded superposition on the electron density surface corresponding approximately to the van der Waals surface. The results for 1,3 dithiane are shown below;
The equivalent calculation for the equatorially substituted mono-sulphoxide is shown below;
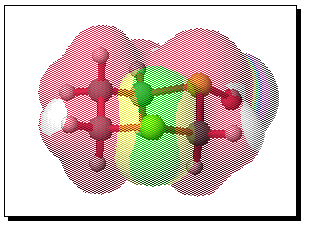
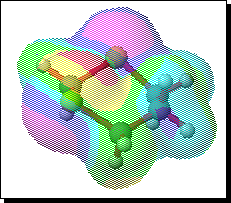
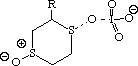
To probe this, we decided to investigate the actual transition states for the oxidation. It is not possible to do this at an ab initio level in a reasonable amount of computation time, and hence we used the much faster semi-empirical PM3 method (Table). A further advantage of the PM3 method is that a good solvation algorithm (COSMO) can be applied to model the polar solvent used in the oxidation using periodate. Transition states could be located for both axial and equatorial oxidations, and the calculated enthalpies for these geometries are shown in the table. For the initial oxidation, the calculations all show a small preference for equatorial attack, consistent with the result obtained from the ab initio calculation of electrostatic potentials. The calculated S...O bond lengths are actually quite short, consistent with a relatively late transition state. The second oxidation again shows little discrimination, with only a slight preference for axial for the system with a t-Bu 5-substituent, well within the error of this type of calculation. This result is again essentially consistent with the previous ab initio calculation. The selectivity shown in the second oxidation step therefore has no apparent explanation within the electrostatic and the solvated transition state models we have investigated.
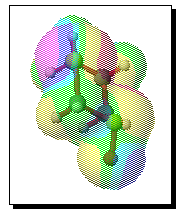
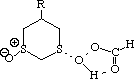
To test the hypothesis that electrophilic oxidation might differ, we also calculated the relevant transition states for oxidation using peroxyformic acid (Table). The PM3 nergies show an equatorial preference for the first oxidation, and again for the second oxidation. This is clearly contrary to the electrostatic argument proposed for the ionic oxidation by periodate, and in fact arises out of HOMO control of the neutral reaction involving peracid. Thus a plot of the HOMO density of 1,3 dithiane superimposed on the van der Waals surface shows clearly that the region of highest coefficient (shown in purple) occurs in the equatorial rather than the axial region, and this is again true for the mono-sulphoxide. As a final test that these influences arise out of stereoelectronic interactions within the 1,3 sulphur system, we carried out calculations on a 1,4 dithiane. Here a marked axial preference for both the first and second oxidations is indicated (Table).
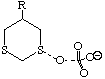 | 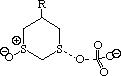
| |||
| eq. | ax. | eq. | ax. | |
| R=H | -430.008 | -429.497 | -470.852 | -469.826 |
| rS-O | 1.603 | 1.610 | 1.601 | 1.609 |
| rI-O | 1.913 | 1.913 | 1.914 | 1.911 |
| R=CH3 | -434.518 | -434.439 | -475.720 | -474.221 |
| rS-O | 1.602 | 1.609 | 1.602 | 1.610 |
| rI-O | 1.914 | 1.913 | 1.915 | 1.914 |
| R=t-Bu | -449.536 | -449.152 | -489.821 | -489.877 |
| rS-O | 1.602 | 1.606 | 1.600 | 1.609 |
| rI-O | 1.914 | 1.912 | 1.915 | 1.911 |
| R=CF3 | -583.854 | -582.683 | -623.770 | -622.516 |
| rS-O | 1.598 | 1.606 | 1.596 | 1.607 |
| rI-O | 1.917 | 1.917 | 1.918 | 1.917 |
| R=NH2 | -425.472 | -423.729 | -465.091 | -465.169 |
| rS-O | 1.601 | 1.608 | 1.599 | 1.608 |
| rI-O | 1.915 | 1.913 | 1.916 | 1.914 |
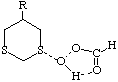 | 
| |||
| eq. | ax. | eq. | ax. | |
| R=H | -91.770 | -90.920 | -131.677 | -130.870 |
| rS-O | 2.599 | 2.589 | 2.594 | 2.577 |
| rH-O | 1.839 | 1.841 | 1.839 | 1.841 |
| rO-O | 1.570 | 1.572 | 1.571 | 1.574 |
| R=t-Bu | -111.679 | -110.464 | -151.318 | -150.610 |
| rS-O | 2.588 | 2.582 | 2.649 | 2.571 |
| rH-O | 1.838 | 1.841 | 1.839 | 1.841 |
| rO-O | 1.571 | 1.573 | 1.564 | 1.575 |
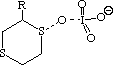 | 
| |||
| eq. | ax. | eq. | ax. | |
| R=t-Bu | -443.643 | -444.195 | -478.250 | -478.504 |
| rS-O | 1.602 | 1.613 | 1.602 | 1.611 |
| rI-O | 1.916 | 1.912 | 1.916 | 1.912 |
This investigation enjoyed the support of the SERC and Pfizer Central Research. We are also particularly grateful to CAChe Scientific for an evaluation copy of the Mulliken program system and to Michael J. Witty of Pfizer Central Research, Sandwich, Kent for helpful discussions. HC thaks JNICT for financial support
REFERENCES
1. Page, P. C. B.; Prodger, J. C.; Hursthouse, M.; Mazid, M. J. Chem. Soc.,Perkin Trans. 1, 1990, 167; Page, P. C. B.; Prodger, J. C. Synlett, 1990, 460; Page, P. C. B.; Prodger, J. C. Synlett, 1991, 84; Page, P. C. B.; Shuttleworth, S. J.; Schilling, M. B.; Tapolczay, D. J. Tetrahedron Lett., 1993, 34, 6947. Page, P. C. B.; Prodger, J. C.; Westwood, D. Tetrahedron, 1993, 49, 10355; Page, P. C. B.; Allin, S. M.; Collington, E. W.; Carr, R. A. E. J. Org. Chem., 1993, 58, 6902; Page, P. C. B.; Allin, S. M.; Collington, E. W.; Carr, R. A. E. Tetrahedron Lett., 1994, 35, 2427; Page, P. C. B.; Allin, S. M.; Klair, S. S.; Collington, E. W.; Carr, R. A. E. Tetrahedron Lett., 1994, 35, 2607; Page, P. C. B.; Shuttleworth, S. J.; McKenzie, M. J.; Schilling, M. B.; Tapolczay, D. J. Synthesis, 1995, 73; Page, P. C. B.; McKenzie, M. J.; Allin, S. M.; Collington, E. W.; Carr, R. A. E. Tetrahedron, 1995, 51, 1285.
2. Page, P. C. B.; Westwood, D.; Slawin, A. M. Z.; Williams, D. J. J. Chem. Soc.,Perkin Trans. 1, 1989, 185; Page, P. C. B.; Westwood, D.; Slawin, A. M. Z.; Williams, D. J. J. Chem. Soc.,Perkin Trans. 1, 1989, 1158; Page, P. C. B.; Klair, S. S.; Westwood, D. J. Chem. Soc.,Perkin Trans. 1, 1989, 2441.
3. Page, P. C. B.; Westwood, D. Unpublished observations.
4. Page, P. C. B.; Wilkes, R. D.; Barkley, J. V.; Witty, M. J. Synlett, 1994, 547; Page, P. C. B.; Gareh, M. T.; Porter, R. A. Tetrahedron: Asymmetry, 1993, 2139; Page, P. C. B.; Heer, J. P.; Bethell, D.; Collington, E. W.; Andrews, D. M. Tetrahedron Lett., 1994, 35, 9629; Page, P. C. B.; Wilkes, R. D.; Witty, M. Org. Prep. Proc. Intl., 1994, 26, 702; Page, P. C. B.; Heer, J. P.; Bethell, D.; Collington, E. W.; Andrews, D. M. Synlett, 1995, in press.
5. Page, P. C. B.; Namwindwa, E. S.; Klair, S. S.; Westwood, D. Synlett, 1990, 457; Page, P. C. B.; Namwindwa, E. S. Synlett, 1991, 80; Barkley, J.; Page, P. C. B.; Dodd, I. M.; Harding, M. M.; Namwindwa, E. S. Acta Cryst (C), 1992, C48, 1689; Barkley, J.; Page, P. C. B.; Dodd, I. M.; Harding, M. M.; Namwindwa, E. S. Acta Cryst (C), 1992, C48, 2039.
6. Cook, M. J.; Tonge, A. P. Tetrahedron Lett., 1973, 14, 849; Cook, M. J.; Tonge, A. P. J. Chem. Soc.,Perkin Trans. 2, 1974, 767; van Acker, L.; Anteunis, M. Tetrahedron Lett., 1974, 15, 225; Khan, S. A.; Lambert, J. B.; Hernandez, O.; Carey, F. A. J. Am. Chem. Soc., 1975, 97, 1468; Bryan, R. F.; Carey, F. A.; Hernandez, O.; Taylor, I. F. J. Org. Chem., 1978, 43, 85; Juaristi, E.; Guzman, J.; Kane, V. V.; Glass, R. S. Tetrahedron, 1984, 40, 1477.
7. Aggarwal, V. K.; Davies, I. W.; Maddock, J.; Mahon, M. F.; Molloy, K. C. Tetrahedron Lett., 1990, 31, 135; Aggarwal, V. K.; Davies, I. W.; Franklin, R. J.; Maddock, J.; Mahon, M. F.; Molloy, K. C. J. Chem. Soc., Perkin Trans. 1, 1991, 662; Aggarwal, V. K.; Davies, I. W.; Franklin, R. J.; Maddock, J.; Mahon, M. F.; Molloy, K. C. J. Chem. Soc., Perkin Trans. 1, 1994, 2363; Aggarwal, V. K.; Evans, G.; Moyo, E.; Dowden, J. J. Org. Chem., 1992, 57, 6390.
8. Aggarwal, V. K.; Davies, I. W.; Franklin, R. J.; Maddock, J.; Mahon, M. F.; Molloy, K. C. J. Chem. Soc., Perkin Trans. 1, 1991, 662; Bien S.; Celebi, S. K.; Kapon, M. J. Chem. Soc., Perkin Trans. 2, 1990, 1987; Auret, B. J.; Boyd, D. R.; Dunlop, R.; Drake, A. F. J. Chem. Soc., Perkin Trans. 1, 1988, 2827; Carey, F. A. Phosphorus Sulphur, 1981, 163; van Acker, L.; Anteunis, M. J. O. Bull. Soc. Chim. Belg., 1977, 86, 299; Johnson, C. R.; McCants, C. M. J. Am. Chem. Soc., 1965, 87, 1109; Bennet, G. M.; Stratham, F. S. J. Chem. Soc., 1931, 1684; Allinger, N. L.; Kao, J. Tetrahedron, 1976, 32, 529.
9. 5-tert-butyl-1,3-dithianes and their sulphoxides have found use as insect control agents: Wacker, V. J.; Toia, R. F.; Casida, J. E. J. Agric. Food Chem., 1992, 40, 497; Wacker, V. J.; Casida, J. E. J. Agric. Food Chem., 1993, 41, 296.
10. Elliott, M.; Pulman, D. A.; Larkin, J. P.; Casida, J. E. J. Agric. Food Chem., 1992, 40, 147.
11. Page, P. C. B.; Klair, S. S.; Brown, M. P.; Smith, C. S.; Maginn, S. J.; Mulley, S. Tetrahedron, 1992, 48, 5933; Page, P. C. B.; Klair, S. S.; Brown, M. P.; Harding, M. M.; Smith, C. S.; Maginn, S. J.; Mulley, S. Tetrahedron Lett., 1988, 29, 4477.
12. Mikolajczyk, M.; Graczyk, P. P.; Wieczorek, M. W. J. Org. Chem., 1994, 59, 1672.
13. Johnson, C. R.; McCants, D. M. J. Am. Chem. Soc., 1964, 86, 2935.
14. Aggarwal, V. K.; Worrall, J. M.; Adams, H.; Alexander, R. Tetrahedron Lett., 1994, 35, 6167.