All the semiempirical calculations were carried out by means of the MOPAC 6.0 package [6]. Transition structures (TS) were located by using the Eigenvector Following algorithm (TS keyword). All the stationary points were fully characterized by testing the sign of the Hessian matrix eigenvalues, and the normal mode vibration corresponding to the only negative eigenvalue of the Hessian, in the case of TS.
In a first approach, given that the 1,3-dipolar cycloadditions are known to have reagent-like transition states, it can be thought that the conformational preference of the dipolarophile is important in order to determine the stereochemical course of the reaction. This hypothesis has already been invoked in the case of the Diels-Alder reaction of 1Z with cyclopentadiene [7].
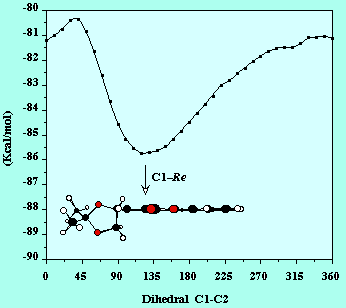
As can be seen in the above Figure , there is only a deep minimum in the rotation of the chiral auxiliary around the C1-C2 bond. This minimum corresponds to a conformation in which the chiral auxiliary shields the C1-Si face of the double bond, which is in agreement with the diastereofacial selectivity observed. However, this is a rather crude approach, since selectivity actually depends on the energy differences of the TS leading to the different products.
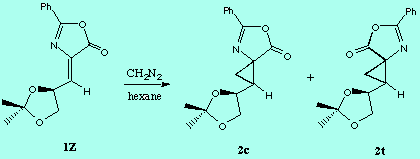
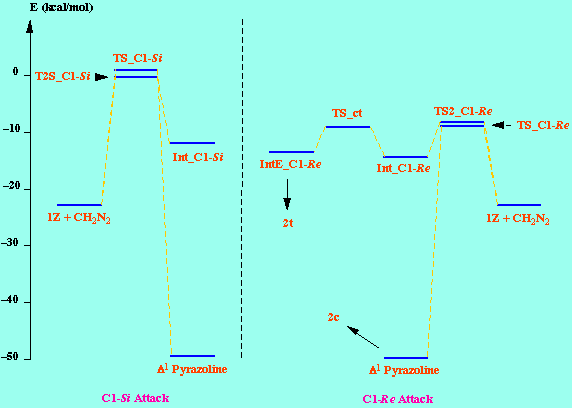
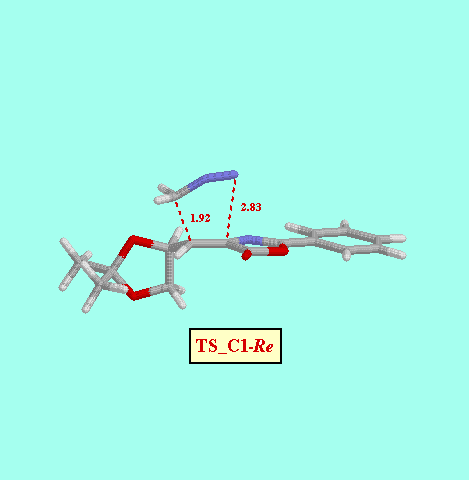
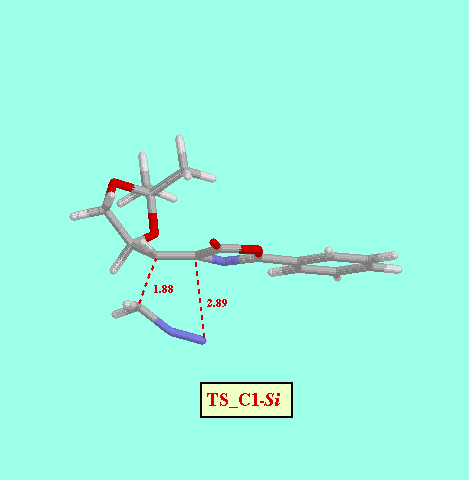
It is generally accepted that 1 pyrazolines are intermediates in the reactions of diazomethane with dipolarophiles to give cyclopropanes. Is some cases (but not in the present case) these pyrazolines can be isolated and characterized [8]. So, in an initial stage we examine the potential energy surfaces (PES) of the addition of diazomethane to 1Z to give the corresponding pyrazolines, by considering both the C1-Re and C1-Si approaches. The resulting PES indicated very asynchronous, but concerted, transition structures (TS_C1-Re and TS_C1-Si), which were further located using the TS keyword of the MOPAC program. The concertedness of this reaction path was confirmed by means of IRC calculations starting from both TS. The energy difference between both TS (9.91 kcal/mol, Table ) indicates an almost total diastereofacial selectivity, the attack of diazomethane proceeding through the C1-Re face of 1Z, in agreement with the observed stereochemistry of the reaction.
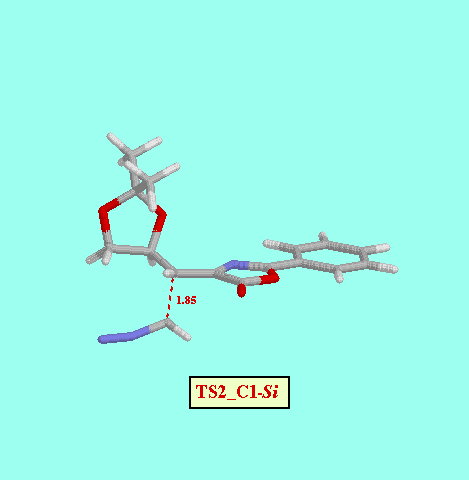
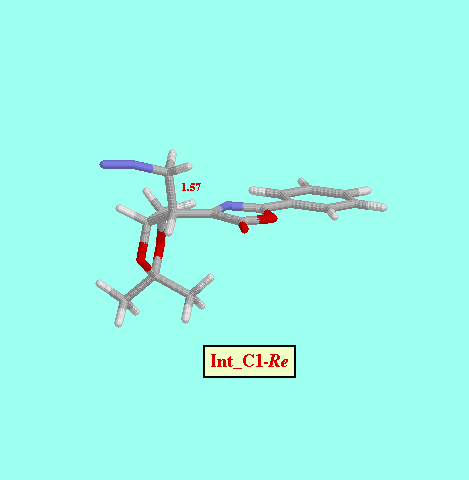
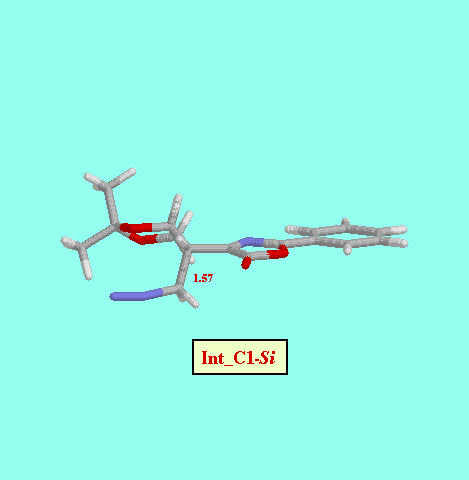
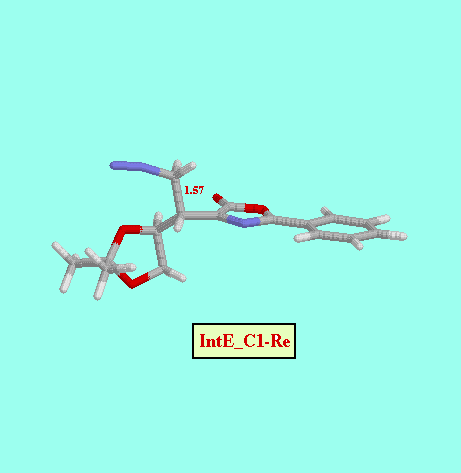
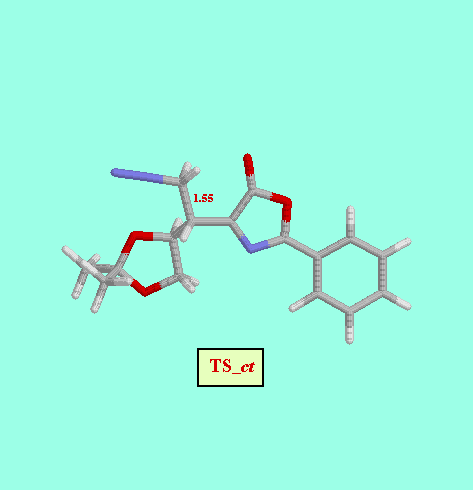
Given that in TS1 the C-N bond is very little formed, we examined a second, not concerted, reaction path, and we foud two more possible TS (TS2_C1- Re and TS2_C1- Si ), as well as two reaction intermediates (Int_C1-Re and Int_C1-Si ). The energy difference between both TS (8.52 kcal/mol, Table ) is also consistent with a high diastereofacial selectivity. However, the existence of non-cyclic intermediates allows the easy interconversion between Z and E isomers of the oxazolone, each isomer leading to different cyclopropanes (cis and trans, respectively). A rotational barrier of 5.20 kcal/mol from Int_C1-Re to IntE_C1-Re has been estimated by locating the corresponding rotational TS (TS_ct ).
The calculated reaction profile is summarized in this Scheme:
[2] (a) C. Cativiela, M. D. Diaz de Villegas, J. A. Mayoral, A. Avenoza, J. M. Peregrina, Tetrahedron, 49, 677 (1993). (b) C. Cativiela, M. D. Diaz de Villegas, A. Avenoza, J. M. Peregrina, Tetrahedron, 49, 10987 (1992).
[3] See for example: (a) S. W. King, J. M. Riordan, E. M. Holt, C. H. Stammer, J. Org. Chem., 47, 3270 (1982). (b) I. Arenal, M. Bernabe, H.. Fernandez-Alvarez, S. Penades-Ullate, Synthesis, 773 (1985). (c) J. Bland, A. Shah, A. Bortolusi, C. H. Stammer, J. Org. Chem., 53, 992 (1988).
[4] For a review see: U. Schmidt, A. Lieberknecht, J. Wild, Synthesis, 159 (1988).
[5] C. Cativiela, M. D. Diaz de Villegas, A. I. Jimenez, F. Lahoz, Tetrahedron Lett., 35, 617 (1994).,
[6] J. J. P. Stewart, MOPAC 6.0, QCPE 455.
[7] E. Buñuel, C. Cativiela, M. D. Diaz de Villegas, J. I. Garcia, Tetrahedron:Asymmetry, 5, 759 (1994).
[8] C. Cativiela, M. D. Diaz de Villegas, J. A. Mayoral, E. Melendez, J. Org. Chem., 50, 3167 (1985).
| Geometry (.pdb file) | Heat of Formation (Kcal/mol) |
|---|---|
| TS_C1-Re | -8.96 |
| TS_C1-Si | 0.95 |
| TS2_C1-Re | -8.36 |
| TS2_C1-Si | -0.16 |
| Int_C1-Re | -14.32 |
| Int_C1-Si | -11.90 |
| IntE_C1-Re | -13.85 |
| TS_ct | -9.12 |
{kind=link}
{kind=link}
{kind=link}
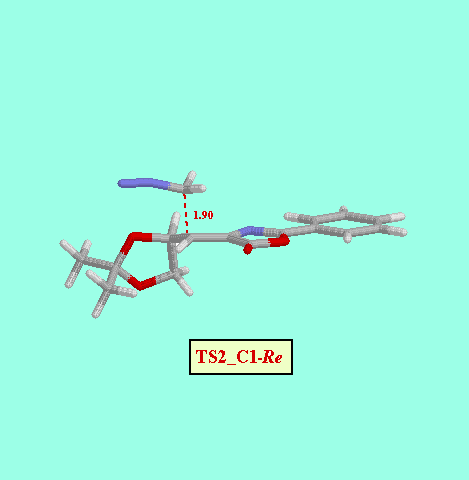{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}