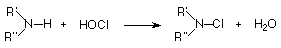
The chemistry of N-halo compounds has scarcely received any attention since Challis and Butler [1] wrote in 1968 "There have been no detailed investigations of the mechanism of amine halogenation...". This has been so even in view of their widely recognized carcinogenic and / or mutagenic properties[2,3]. These compounds are generated during water disinfection with halogens, and under different conditions give place to several other toxic substances through different pathways (disproportionation[4,5], Grob fragmentation[6] and inter- and intramolecular (1,2)-elimination reactions[7,8] have been identified).
The chlorination of amines, amino acids and peptides by HOCl (the most commonly used water-disinfecting agent) to yield the N-Cl-compounds has been shown [9,10,11] to proceed via direct transfer of Cl from the O of the HOCl to the unprotonated N of the N-compound.
Scheme 1.
Notwithstanding the few recent studies available, some uncertainties still remain about this mechanism, one of the most important being whether the reaction is difussion-controlled or not. Here, we present some evindences to discard the difussion-controlled process in favour of a chemically controlled one.
Experimental.
Reagents. Commertial amino compounds of the best quality available were used without further purification. Aqueous chlorine solutions were used as chlorinating agent. These were generated every 3 or 4 days by bubling Cl2 (g) through a NaOH solution and spectrophotometrically titrated daily (λmax(H2O)=292 nm, ε=350 dm3.mol-1.s-1 for ClO- when pH>12)[12]. CH3COOH / CH3COO-, H2PO4- / HPO4-2, H3BO3 / NaOH and HCO3-/ CO3-2 buffers, as well as NaOH solutions were used to control the pH. The total buffer concentration was kept about 0.02 mol.dm-3 in all cases and the pH>5 to avoid the presence of Cl2 (aq). Ionic strength was kept constant to 0.5 mol.dm-3 using NaClO4.
Equipment. SF-61 single-mixing and SF-61MX multi-mixing stopped flow spectrophotometers from Hi-Tech Scientificreg. were used to follow the reaction. This was done either by measuring the increase in absorbance due to the formation of the N-Cl-compounds or the decrease due to the consumption of the chlorinating agent. The temperature was kept constant to within +/-0.05 K by means of water-flow thermostating devices. The pH measurements were carried out with previously calibrated combined glass electrodes. The achieved accuracy for pH was +/-0.02 units.
Working procedure. All reactions were carried out under second order conditions, i.e.: by simultaneous mixing of equal concentrations of both the chlorinating agent and the N-compound. The integrated second order rate equation was adequately fitted to the A/t data using the Marquardt non-linear optimization algorithm[13]. Each rate constant reported here is an average of, at least, five kinetic runs, the reproducibility being better than 4 %.
Discussion.
The chlorination of amino compounds is a second order reaction, one order relative to each of the reagents:
The rate constant changes dramatically with the acidity of the medium, passing through a maximum at a pH corresponding to the average of the pKa's of both reagents (Figure 1).
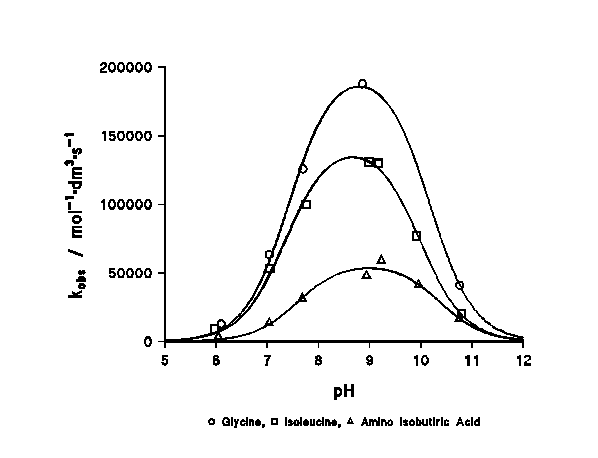
Figure 1: profile of the reaction rate with the pH. The mechanism for this reaction has been previously discussed elsewhere9, concluding that the reaction proceeds as shown in Scheme 1 (vide supra). The theoretical expression deduced for the observed rate constant is:
where k is the bimolecular rate constant for the process and Ka and Kc are the ionization constants for the N-compound and for HOCl, respectively. By fitting the experimental data to this equation we have worked out the bimolecular rate constants for a wide variety of primary and secondary N-compounds, including amines, amino acids, peptides, chloramines and amides.
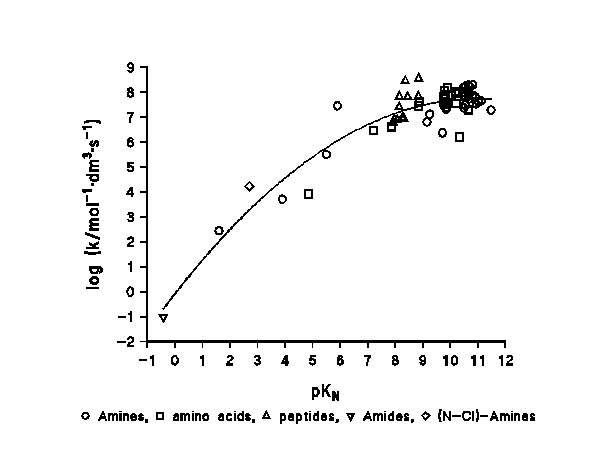
Figure 2: reactivity - basicity / nucleophilicity relationship in the chlorination of N-compounds.
A plot of [log (k/mol-1dm3s-1)] versus the pKa of the N-compound shows a clear relationship between the basicity / nucleophilicity of the N-compounds and their reactivity towards the chlorinating agent. This behaviour has been found in a range of 13 pK units for more than 70 compounds, as shown in Figure 2.
In view of this behaviour, it could be considered that when pKN>9.0, then:
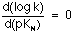
and the reaction has reached the difussion-control limit. This hypothesis, that has been pointed out previously[14], could be reinforced by the values we have obtained for the activation enthalpies of this reaction once accounted for the acid-base equilibria of both reagents (Table 1).
| N-Compound | ΔH / kJ.mol-1 | ΔS / J.mol-1.K-1 |
|---|---|---|
| Me2NH | 15 +/- 5 | -46 +/- 6 |
| Me,EtNH | 23 +/- 3 | -18 +/- 2 |
| Et2NH | 26 +/- 6 | -11 +/- 1 |
| Pr2NH | 29 +/- 2 | -2 +/- 0.2 |
| iPr2NH | 30 +/- 4 | -5 +/- 1 |
| iBu2NH | 26 +/- 6 | -18 +/- 2 |
| Iminodipropionitrile | 10 +/- 1 | -108 +/- 2 |
| Gly.Gly | 0 | -101 +/- 20 |
| Gly.Gly.OEt | 0 | -131 +/- 15 |
I.e.: the very small ΔH could correspond to a difussion-controlled process, although ΔS lies in a wider range, even for compounds that have very similar pKN values and for which pKN > 9.0.
Notwithstanding these facts, there seems to be enough evidence to discard a difussion-controlled mechanism:
a) The higher rate constants we have found are around 108-109 mol-1.dm3.s-1, still well below the generally accepted limit of difussion-control. Table 2 summarizes the rate constants for the chlorination of Me2NH by different chlorinating agents, showing that there is no apparent relationship between the reaction rate and the size of the chlorinating agent, as should occur if the reaction were difusion-controlled.
| Chlorinating agent | k /mol-1.dm3.s-1 |
|---|---|
| (N-Cl),(N-Me)-Benzenesulfonamide | 0.0245.107 |
| (N-Cl)-Succinimide | 1.53.107 |
| (N-Cl)-Quinuclidinium | 2.019.107 |
| HOCl | 7.501.107 |
| NH2Cl | 80.107 |
| Cl2 | 160.107 |
is very likely to take place and would be thermodinamically favoured. The molecules under study are quite polar and we have no way to account for the influence of the changes in solvation upon the activation parameters.
c) We have found a clear steric effect within a family of similar compounds, the reaction rate decreasing as the steric hindrance increases. Figure 3 shows this effect using Charton's ν parameter. The effect is found both for the groups directly attached to the N (case of the amines) and for those attached to a C in the vicinity of the N (case of the amino acids), being more important in the first case. This behaviour does not agree with which would be expected for a physically-controlled reaction, but with a chemically-controlled one.
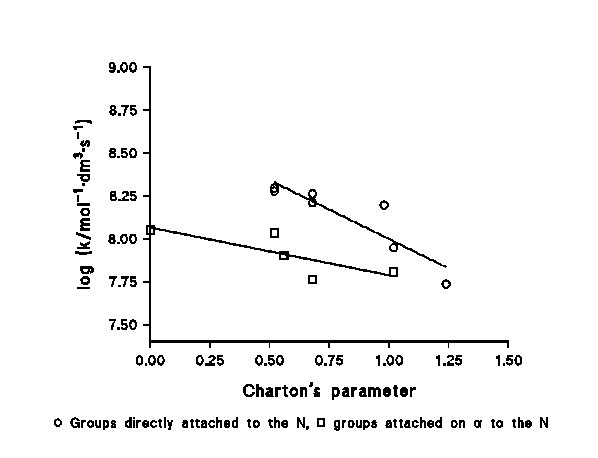
Figure 3: Steric effects in the chlorination of N-compounds by HOCl.
d) We have found values of (kH/kD)<1 for the solvent isotope effect in the chlorination of several N-compounds in the pKN range 3.90<pKN<12.4 by HOCl. Thus, for example, (kH/kD)=0.70 for Glycine (pKN=9.78). Again, this behaviour would not be expected for a difussion-controlled process.
Conclusion.
Taking into account all the previous evidences, we come to the conclusion that the chlorination reaction through transfer of Cl from the O of HOCl to the N of the corresponding N-compound is a chemically controlled process, at least in the wide range of pKN we have studied.
Work in progress.
In view of the very special reactivity - basicity / nucleophilicity pattern observed, we are re-visiting this mechanism in terms of a halogen transfer reaction, as compared with other well-known group transfers. We are also carrying out more research in order to characterize the transition state.
References.
[1] B. Challis, A.R. Butler. "Substitution at an amino nitrogen", in "The Chemistry of the Amino Group". Edited by S. Patai. Interscience Publishers. London, 1968.
[2] J. Owusu-Yaw, W.B. Wheeler, C.I. Wei. Water Chlorination. Environmental Impact and Health Effects. Vol. 6, pp. 179-191. Lewis Publishers. Inc. Chelsea, 1990.
[3] A.C. Sen, J. Owusu-Yaw, W.B. Wheeler, C.I. Wei. J. Food. Sci., 54 (4), 1057-1060&1065 (1989).
[4] J.M. Antelo, F. Arce, M. Parajó, P. Rodríguez, A. Varela. Int. J. Chem. Kinet., 24, 991-997 (1992).
[5] X.L. Armesto, M. Canle L., M. Losada, J.A. Santaballa. Int. J. Chem. Kinet., 25, 331-339 (1993).
[6] X.L. Armesto, M. Canle L., M. Losada, J.A. Santaballa. J. Org. Chem., 59, 4659-4664 (1994).
[7] X.L. Armesto, M. Canle, M. Losada, J.A. Santaballa. J. Chem. Soc., Perkin Trans. 2, 181-185 (1993).
[8] X.L. Armesto, M. Canle L., P. Carretero, M. Losada, J.A. Santaballa. Unpublished work.
[9] X.L. Armesto, M. Canle L., J.A. Santaballa. Tetrahedron, 49, 275-284 (1993).
[10] X.L. Armesto, M. Canle L., M.V. García, M. Losada, J.A. Santaballa. Int. J. Chem. Kinet., 26, 1135-1141 (1994).
[11] X.L. Armesto, M. Canle L., M.V. García, M. Losada, J.A. Santaballa. Gazz. Chim. Ital., 124, 519-523 (1994).
[12] J.C. Morris. J. Phys. Chem., 70, 3798 (1966).
[13] D.W. Mardquardt. J. Soc. Ind. Math., 11, 431 (1963).
[14] D. Matte, B. Solastiouk, A. Merlin, X. Deglise. Can. J. Chem., 67, 786-791 (1989).