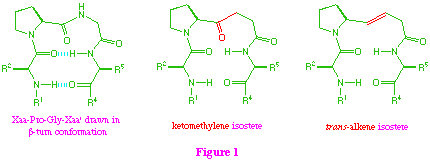
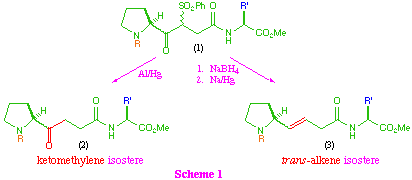
Our synthetic approach to these isosteres proceeds through a common beta-ketosulfone intermediate (1) which on direct reduction of the sulfone group provides the ketomethylene isostere (2), and also allows access to the trans-alkene system (3) by initial reduction of the keto-function, followed by reductive elimination (scheme 1).
From a strategic point of view, when applying this approach to tetra-peptide isosteres, the additional amino acid residue (R = L-alanine) can be incorporated at the start of the synthesis, or can be introduced at the end by coupling with the proline residue. Since the two alternative strategies both have potential problems, we decided to investigate them both.
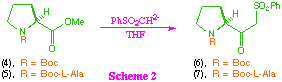
First we examined synthetic routes to the key beta-ketosulfone intermediate (1). This required the initial synthesis of two proline derived beta-ketosulfones (5) and (6), which was attempted via reaction of the corresponding methyl esters (4) and (5) with the dianion derived from methylphenylsulfone (scheme 2). As anticipated based on our previous studies, under these reaction conditions, N-t-butoxycarbonyl-L-proline methyl ester (4) gave the desired beta-ketosulfone (6) in good overall yield, although the product was contaminated with small amounts (>5%) of methylphenyl sulfone. We were surprised however, to find that the dipeptide (5) also gave the corresponding beta-ketosulfone (6) in reasonable yield, with no sign of epimerisation. Given the number of acidic sites in the substrate (5), the relative success of this transformation clearly demonstrates the powerful nucleophilicity of the sulfone derived dianion.
It was also necessary to prepare amino acid derived bromoacetates (10) and (11), which were to be used in the construction of the peptide isosteres. This was readily achieved by acylation of the corresponding amino acid ester hydrochlorides (8) and (9) as outlined in scheme 3[6].

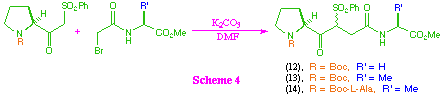
Coupling of the beta-ketosulfones with the bromoacetates to give tri- and tetra-peptide systems (12)-(14) was then investigated (scheme 4). These couplings were effected by the use of anhydrous potassium carbonate in dry N,N-dimethylformamide, and gave the corresponding products in good yield. In all cases the compounds were isolated as a mixture of diastereoisomers at the newly created chiral centre, but showed no evidence of epimerisation at other sites.
Compounds (12) and (13) were readily desulfonylated with aluminium amalgam, to give the ketomethylene tri-peptide isosteres (15) and (16) (scheme 5), however substrate (14) was recovered unchanged from the same reaction conditions. This observation is consistent with previous work in our group which suggests that the aluminium amalgam desulfonylation tends to be unsuccessful when applied to sterically hindered substrates, and to date we have been unable to find an effective method for the desulfonylation of (14).
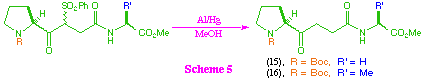
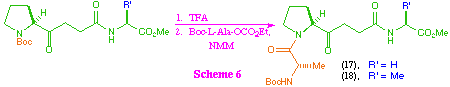
As a consequence of being unable to access the tetra-peptide ketomethylene isosteres (17) and (18) directly from (5), we investigated the possibility of their synthesis from the tri-peptide systems (15) and (16) (scheme 6). After investigation of a range of coupling protocols, it was found that this could most effectively be achieved by removal of the t-butoxycarbonyl protecting group to give the corresponding amine salt, and then coupling with the mixed anhydride of N-protected L-alanine in the presence of N-methylmorpholine. Under these reaction conditions, the tetra-peptide ketomethylene isosteres were formed despite the obvious problems associated with the generation of a free amine in the presence of a keto-function.
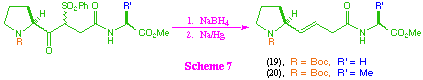
The attempted conversion of beta-ketosulfone intermediates (12)-(14) to the corresponding trans-alkene isosteres was less successful (scheme 7). Substrates (12) and (13) did give the desired products (19) and (20), but in relatively poor overall yield, and again the intermediate (14) failed to give any of the desired product. The main problems encountered in this transformation were competing reduction of the methyl ester group on treatment with sodium borohydride, and over-reduction of the alkene product to the corresponding saturated system. In addition, it was found that the product was readily transesterified in the presence of an alcohol, including transesterification to the corresponding ethyl ester if ethyl acetate was used in the reaction work up. These facile transesterifications are partly a function of the structure of the products, and partly a consequence of the highly basic nature of the sodium amalgam reduction process. The use of disodium hydrogenphosphate as buffer in this reaction system is only partialy successful due to the heterogeneous nature of the reaction system, and alternative buffers were equally unsuccessful.
Despite the disappointing yields for the reductive desulfonylation, we did examine the utility of this type of intermediate in the formation of tetra-peptide systems (scheme 8). Thus coupling of the tri-peptide trans-alkene isostere (21) with the mixed anhydride derived from N-protected L-alanine, under the same conditions as used for the ketomethylene systems, gave the desired tetra-peptide system (22), in comparable yield.

In conclusion, we have developed a synthetic approach to a series of tri- and tetra-peptide analogues which incorporate a modified Pro-Gly linkage. We are currently in the process of developing improved procedures for the preparation of the trans-alkene system, and in studying the conformational preferences of these novel systems.
Acknowledgements
We would like to thank Mrs. R. Howard for mass spectra.
Experimental - General
Melting point determinations were carried out on an electrothermal apparatus and were recorded uncorrected. Optical rotations were measured on an AA-10 monochromatic 589 nm (Optical Activity Ltd.) polarimeter at room temperature. Infra-red absorption spectra were run neat on a Pye Unicam SP3-100 IR spectrophotometer. Proton nmr spectra were recorded at 300MHz on a Bruker AC-300 instrument, as solutions in either deuteriochloroform or d6-dimethylsulfoxide. Chemical shifts are referenced to tetramethylsilane, and J-values are rounded to the nearest 0.5Hz. Mass spectra were recorded using ammonia chemical ionisation at low resolution on a Finnigan 4500 instrument, and at high resolution on a Kratos Concept 1-S instrument. Thin layer chromatography was carried out using Merk Kieselgel 60 F254 glass-backed plates. The plates were visualised by the use of a UV lamp, or by dipping in a solution of vanilin in ethanolic sulfuric acid, followed by heating. Silica gel partical sizes 40-63microm was employed for flash chromatography. All solvents were dried using standard procedures.