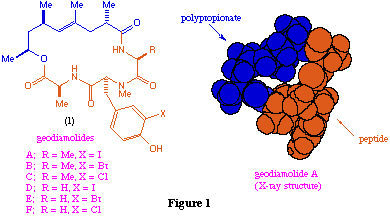
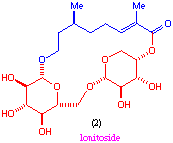
Our interest in these materials lies in the unusual structural combination of the hydrophobic polypropionate and polar peptide chains (fig. 1) and from the fact that related features are also found in other natural products such as lonitoside[7] (2) which is a macrocyclic lactone composed of carbohydrate and terpene fragments.
We are interested in the possibility of constructing novel biologically-active macrocycles which incorporate polar and non-polar fragments similar to those found in such natural products, and in this paper report synthetic approaches to two tri-L-alanyl derived macrocyclic systems related to the geodiamolides.
Initially we chose to examine the terpenoid (S)-citronellal (3) as a starting material for the synthesis of polypropionate-like fragments, and this was achieved as outlined in scheme 1. Addition of methylmagnesium bromide to citronellal, followed by protection of the resulting hydroxyl group as a tert-butyldimethylsilyl ether gave intermediate (4) as a 1:1 mixture of diastereoisomers. Although these diastereoisomers could be separated by column chromatography, this was not done as we were interested in generating a range of isomers of the final macrocycles. Ozonolysis of the alkene moiety in (4), followed by Wittig reaction, then gave compound (5). This material was readily converted into the polypropionate-like fragment (6) by hydrogenation followed by saponification. Again as expected, the hydrogenation was not stereoselective, and so the product (6) was obtained as a roughly equal mixture of all four diastereoisomers.
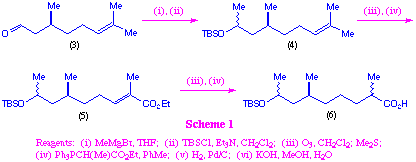
We were interested in using the diastereoisomeric mixture directly in the macrolide formation mainly because this would allow us to determine if there was a kinetic preference for certain diastereoisomers to undergo cyclisation in preference to others. In addition, the use of this diastereoisomeric mixture would allow us to develop a macrocyclisation strategy that is likely to be general for a wide range of polypropionate-like systems.
We investigated a number of methods for the coupling and macrocyclisation of the acids (6) with N-tert-butoxycarbonyl-L-alanyl-L-alanyl-L-alanine methyl ester (7), and found that this was most readily achieved using DCC/HOBT for the initial amide formation, followed by macrolactonisation using BOP-Cl[8] (scheme 2).
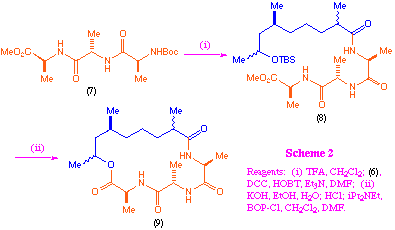
We found no evidence of kinetic selectivity in the macrolactonisation process and the final products (9) were, as far as we could determine, still a roughly equal mixture of four diastereoisomers.
The scheme outlined above represents an effective means of preparing a number of macrocyclic systems which incorporate both tripeptide and a polypropionate-like fragments, however it is not straightforward to extend this methodology to polypropionate systems that incorporate unsaturation at C-4, such as that found in the geodiamolides. In order to prepare such systems we used an alternative approach which employs Wittig chemistry to install this unsaturation (scheme 3).
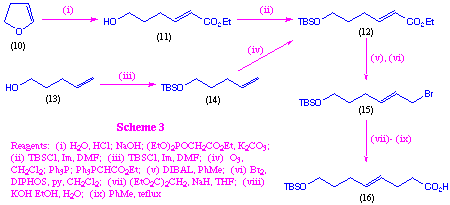
This could readily be achieved using either dihydrofuran (10) or 5-hydroxypentene (13) as the starting material. In the case of dihydrofuran, it was found that hydration to the corresponding lactol, followed by in situ reaction with triethylphosphonoacetate gave intermediate (11) in reasonable overall yield. This one-pot operation proved more efficient than the alternative two-step processes, particularly on large scale. Protection of the resulting hydroxyl as a tert-butyldimethylsilyl ether then gave the key intermediate (12). This compound was prepared from 5-hydroxypentene (13), by first protecting as the silyl ether, and then via ozonolysis, followed by in situ reaction of the resulting aldehyde with carboethoxymethylenetriphenyl phosphorane. This second approach gave better overall yields of the intermediate (12), but suffers from the disadvantage of utilising more expensive starting materials. Compound (12) was then reduced to the corresponding allylic alcohol using DIBAL, and this was efficiently converted into the allylic bromide (15) via treatment with DIPHOS and bromine[9]. At this stage we examined a variety of methods for the conversion of the bromide (15) into the desired carboxylic acid (16). It was found that direct reaction with ester enolates was not particularly effective, and that this transformation could most readily be achieved by initial reaction with diethyl malonate, followed by hydrolysis and decarboxylation. This gave the parent C-4 unsaturated hydroxy acid in a protected form suitable for incorporation in the macrocyclic systems.
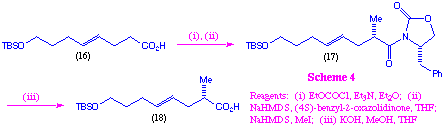
We also decided to examine the use of Evans enolate chemistry as a means of stereoselectively introducing an C-2 methyl substituent into this system (scheme 4). This was achieved via coupling of the (4S)-benzyl-2-oxazolidinone anion with the mixed anhydride derived from acid (16), followed by in situ alkylation with methyl iodide. As anticipated, high diastereoselectivity (96%ee) was obtained in the alkylation step, and hydrolysis then gave the substituted acid (18).
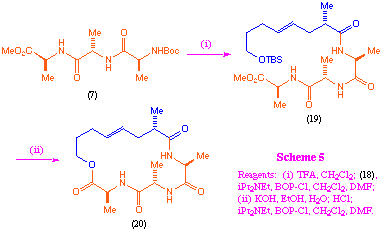
Incorporation of fragment (18) into the macrocyclic unit was achieved in a similar way to that performed for the earlier polypropionate-like systems, however in this case it was found that the initial coupling between (18) and the tripeptide portion was also best achieved using BOP-Cl (scheme 5).
In conclusion, we have developed two approaches to polypropionate-like fragments related to those found in the geodiamolide family of natural products, and successfully used products derived from these in the construction of novel macrocyclic systems. The chemistry developed should be applicable to the synthesis of a wide range of novel peptide-polypropionate macrocycles, and this will be the subject of future investigations.
Acknowledgements
We are grateful to the SERC and Glaxo, Greenford for financial support (CASE studentship to LW). We also thank Dr. C. Smith, for helpful discussions, Dr. M. Stuckey for NMR spectra and Mrs. R. Howard for mass spectra.
Experimental - General
Melting point determinations were carried out on an electrothermal apparatus and were recorded uncorrected. Optical rotations were measured on an AA-10 monochromatic 589 nm (Optical Activity Ltd.) polarimeter at room temperature. Infra-red absorption spectra were run neat on a Pye Unicam SP3-100 IR spectrophotometer. Proton nmr spectra were recorded at 300MHz on a Bruker AC-300 instrument, as solutions in either deuteriochloroform or d6-dimethylsulfoxide. Chemical shifts are quoted on the delta scale and referenced to tetramethylsilane, and J-values are rounded to the nearest 0.5Hz. Mass spectra were recorded using ammonia chemical ionisation at low resolution on a Finnigan 4500 instrument, and at high resolution on a Kratos Concept 1-S instrument. Microanalysis were performed by the microanalytical laboratory at Glaxo Greenford Reseach, Greenford. Thin layer chromatography was carried out using Merk Kieselgel 60 F254 glass-backed plates. The plates were visualised by the use of a UV lamp, or by dipping in a solution of vanilin in ethanolic sulfuric acid, followed by heating. Silica gel partical sizes 40-63um was employed for flash chromatography. All solvents were dried using standard procedures.
Reactions were routinely carried out under an inert atmosphere of argon or nitrogen. Where necessary, solvents and reagents were dried and purified according to recommended methods.[10] Petroleum ether refers to the fraction boiling in the range 40-60degC. Organic solutions were routinely dried over magnesium sulphate and evaporation / concentration refers to solvent removal on a rotary evaporator under reduced pressure. n-Butyllithium (in hexanes) was supplied by Lithco Corporation and was standardised by titration with diphenylacetic acid.