 Figure 1. (enlarged GIF,
16K)
Figure 1. (enlarged GIF,
16K)
The products obtained from the the microwave reactions were identical to
those obtained by classical reflux methods.
The yields in forming the adduct compared well with the reflux method,
and ranged from 55% to as high as 81% which compares to 60% as reported
previously (1).
However, the hydrolysis to form the diamine generally gave yields
under 50%. Using the original conditions of 70% aqueous sulfuric acid,
charring was often evident and so the acid strength was diluted to 50%.
This necessitated extending the reaction time.
The lower yields of the diamine could possibly be due to
incomplete hydrolysis of the adduct.
The times used for the hydrolysis reactions were between three and four
minutes, except for the p-chlorostilbenediamine when the reactants were
irradiated for five minutes.
N-benzoyl-N'-benzylidene meso-1:2-diphenylethylenediamine.
Benzaldehyde (15.1cm3, 15g, 140 mmol) and
ammonium acetate (30.12 g, 390 mmol) were added to a Teflon bomb which was
sealed and placed in a microwave oven.
The vessel was heated for three minutes, cooled and the mixture filtered.
The product was washed with ethanol, air dried then recrystallized
from toluene. M.P. 253-255 C (lit. 258-259 C). Yield 14.32g, 56%. The Mass Spectrum showed m/e at 404.
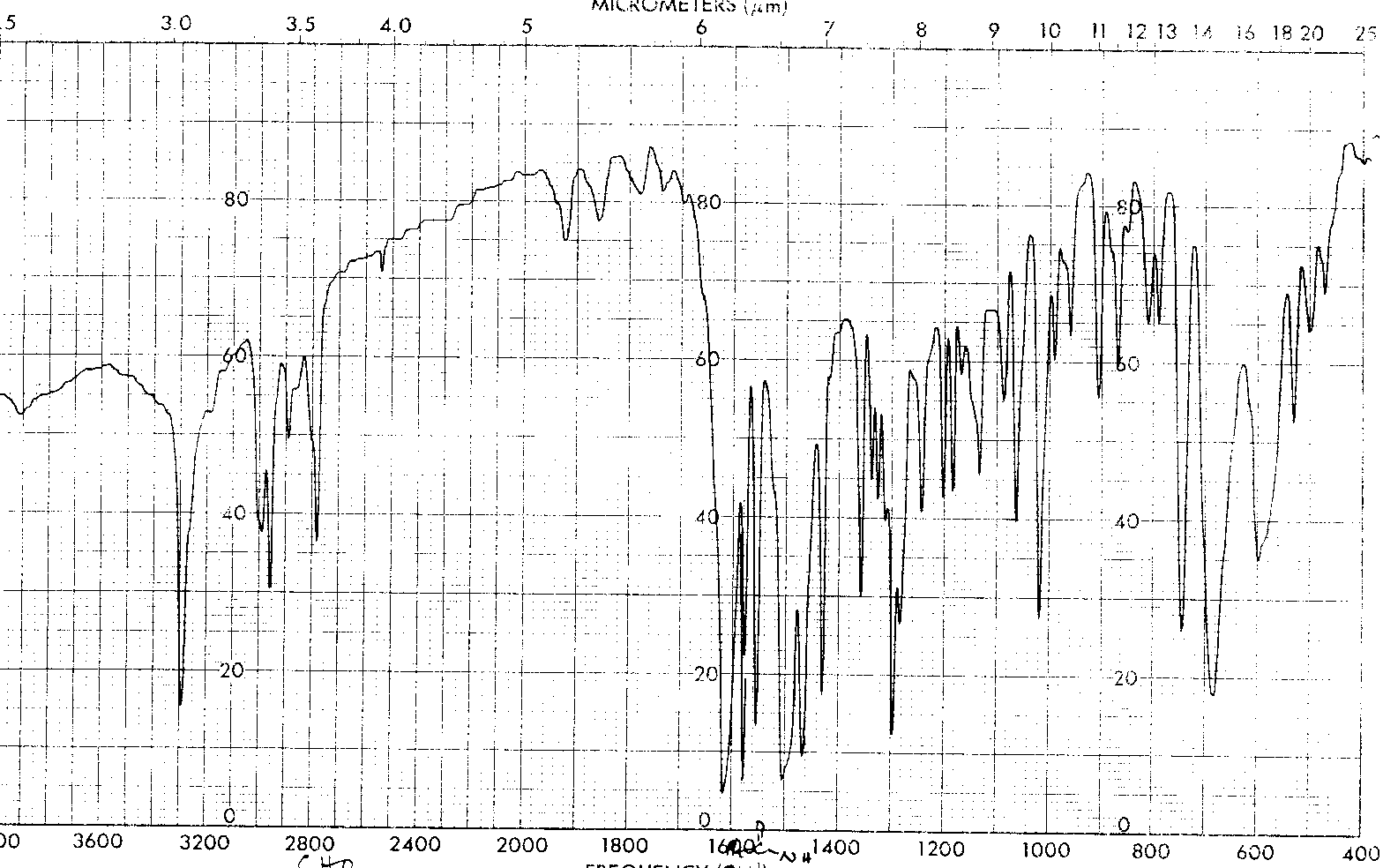
IR Spectrum (56 K GIF file).
The 200 MHz proton NMR (85K JCAMP-DX file) gave signals at 8.30 (s),7.79-7.24 (m), 6.71 (d) 5.55 (dd), 4.95 (d) and 1.55 (s).
meso-1:2-diphenylethylenediamine.
The adduct (5g, 12 mmol) along with 50% sulfuric acid (25 cm3) were
added to a Teflon bomb and placed in a microwave oven. The
vessel was heated for 3.5 minutes and then cooled. The mixture was
extracted with ethyl acetate (3 times 30 cm3). The organic layer, which
contained unreacted starting material and other impurities was discarded.
The aqueous layer was made alkaline (pH~10) using KOH and further
extracted with ethyl acetate. The organic extract was washed
with water, dried with sodium sulfate and filtered.
The filtrate was evaporated in vacuo. The product was recrystallized
from methanol. M.P. 119-120 C (lit.120.5 121.5 C). Yield 2.62g, 24%.
The Mass Spectrum showed m/e at 212.
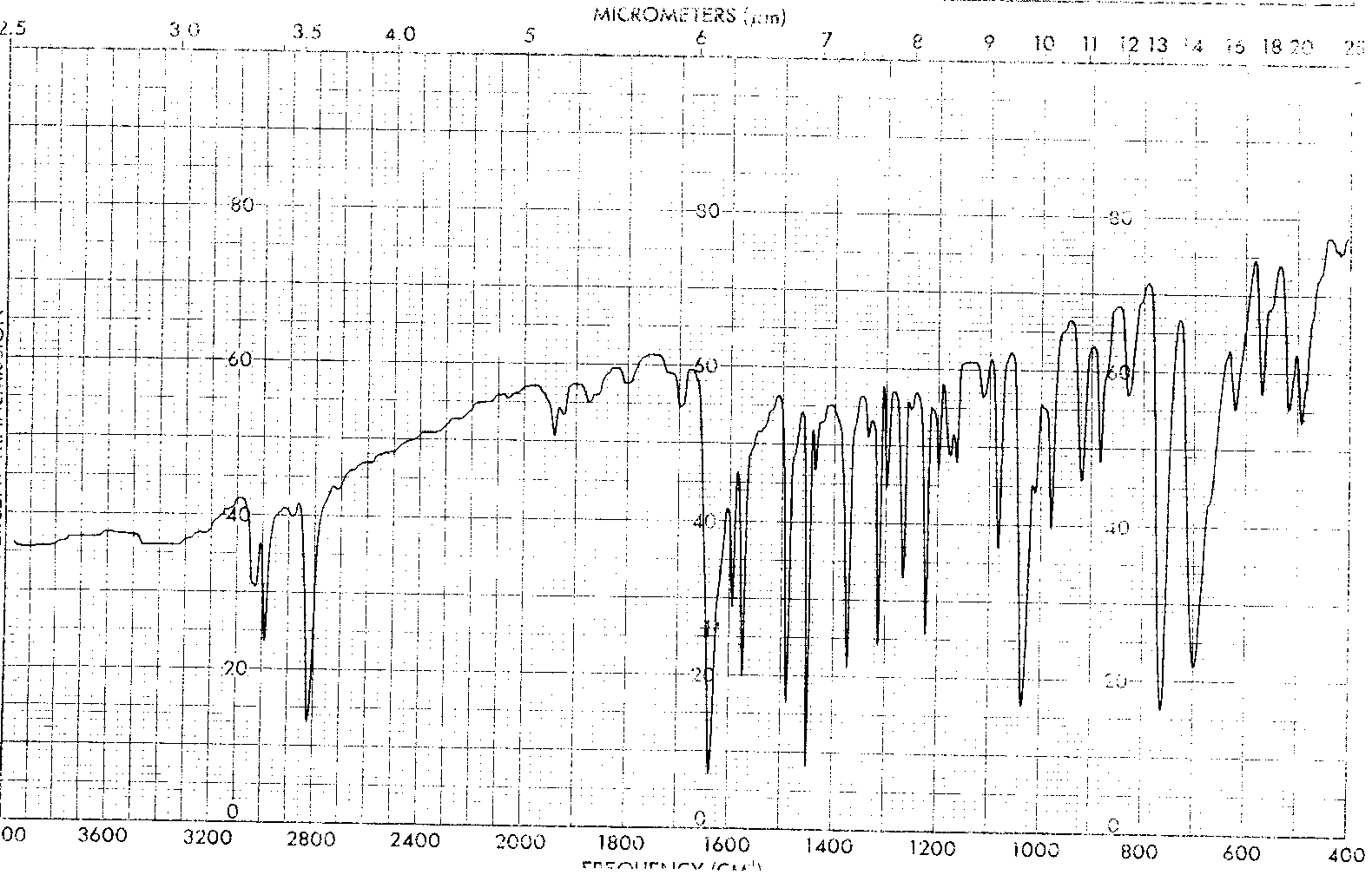
IR Spectrum- (53K GIF file)
The 200 MHz proton NMR (85K
JCAMP-DX file) gave signals at 7.41 (m), 5.41 (s) and 1.47
(s).
Preparation of adduct from p-chlorobenzaldehyde.
p-Chlorobenzaldehyde (5.04 g, 36 mmol) and ammonium acetate (10.06 g, 130
mmol) were added to a Teflon bomb which was sealed and
placed in a microwave oven. The vessel was heated for 2.5 minutes,
cooled and the mixture filtered. The product was washed with ethanol and
air dried. The product was recrystallized from butan-l-ol. M.P.
251-253 C (lit. 249 C). Yield 1.5g, 31%.
The 200 MHz proton NMR (85K
JCAMP-DX file) gave signals at 8.26 (s), 7.77-7.12 (m), 6.62 (d),
5.52 (dd), 4.96 (d), 1.69 (broad-s) and 1.28 (s).
meso-1:2-di-(p-chlorophenyl)ethylenediamine.
The adduct (0.9 g, 2 mmol) and 40% sulfuric
acid (4.5 cm3) were added to a Teflon bomb which was sealed and placed in a
microwave oven. The vessel was heated for 5 minutes,
cooled and the mixture extracted with ethyl acetate (3 times 30 cm3)
which was discarded. The aqueous portion was made alkaline (pH~9) using KOH
and further extracted with ethyl acetate. The organic extract was washed
with water, dried with sodium sulfate and filtered. The filtrate was
evaporated using a rotary evaporator and the product recrystallized from
methanol. M.P. 140-141 C (lit 137-138 C). Yield 0.24g, 51%.
The 60 MHz proton NMR (8K
GIF file) gave signals at 7.25 (Ar-H), 3.9 (N-C-H) and 1.5 (N-H).
Preparation of adduct from m-nitrobenzaldehyde.
m-Nitrobenzaldehyde (5.14 g, 34 mmol) and ammonium acetate
(10.14 g, 132 mmol) were added to a Teflon
bomb which was sealed and heated in a microwave oven for 3 minutes.
The vessel was then cooled and the mixture filtered. The crystals were
washed with ethanol and the product recrystallized from butan-l-ol. M.P.
300 C (sublimes) Yield 4.01g, 81%.
meso-1:2-di-(m-nitrophenyl)ethylenediamine.
The adduct (4.01 g, 7 mmol) and 50% sulfuric acid (23 cm3)
were added to a Teflon bomb which was sealed and heated in a microwave
oven for 4 minutes. The vessel was then cooled and the organic portion
extracted with ethyl acetate (3 times 30 cm3) which was then discarded.
The aqueous portion was made alkaline (pH~10) using KOH and further
extracted with ethyl acetate. The organic extract was washed with water,
dried with sodium sulfate and filtered. The filtrate was evaporated under
reduced pressure and the product was recrystallized from methanol. M.P.
184-187 C (lit. 189-190 C). Yield 0.63g, 30%.
{kind=link}
{kind=link}
{kind=link}