{kind=link}
t/min E/mol% Z/mol%
_______________________________________________________________________________
Fieser mixture* (b.p.: app. 150 oC)
35 79.9 21.1 _______________________________________________________________________________
H2O (b.p.: 100 oC, mu(25): 1.82)
180 94.4 5.6 _______________________________________________________________________________
diethyl ether (b.p.: 35 oC, mu(25): 1.87)
180 100 0 _______________________________________________________________________________
dibutyl ether (b.p.: 142-143 oC, mu(20): 1.15)
40 100 0 _______________________________________________________________________________
diglyme (b.p.: 162 oC, mu(25): 1.97)
180 100 0 _______________________________________________________________________________
benzene (b.p.: 80 oC, mu(20): 0.00)
180 100 0 _______________________________________________________________________________
xylene (isomeric mixture) (b.p.: 138-142 oC, mu(20): 0.30)
30 100 0
180 98.1 1.9
360 97.9 2.1 _______________________________________________________________________________
decalin (isomeric mixture) (b.p.: 192 oC, mu(25): 0.00)
35 94.2 5.8
360 87.4 12.6
540 90.5 9.5
===============================================================================
*Fieser mixture: benzaldehyde, phenylacetic acid, acetic anhydride, triethylamine
The results of thermal as well as photoinduced isomerization reactions revealed that starting from the E isomer an equilibrium mixture of nearly 1:1 composition could be achieved (Table 3). It is true that reaching this composition requires considerable amount of time.
Table 3
Double bond isomerization of E-alpha-phenylcinnamic acid under thermal conditions (reflux in a mixture of acetic anhydride and triethylamine) and upon UV irradiation (in diethyl ether)
=======================================================================
time/h thermal UV light
___________________ ____________________
E Z E Z
_______________________________________________________________________
0 100 0 100 0
22 - - 61.1 38.9
24 96.7 3.3 - -
30 - - 61.1 38.9
48 83.3 16.7 - -
96 66.2 33.6 - -
120 54.7 45.3 - -
168 54.7 45.3 - -
=======================================================================
Computational results
Semiempirical calculations on the isolated molecules gave similar stability data for the two isomers. However, calculations also revealed that the dipole moments were different (Table 4).
Table 4
The standard heat of formation (in kJ/mol) and dipole moment (D in Debye) data for the alpha-phenylcinnamic acid isomers calculated by semiempirical quantum chemical methods
===========================================================================
MNDO AM1 PM3
________________ _________________ __________________
deltaHf,298 D deltaHf,298 D deltaHf,298 D
___________________________________________________________________________
E -109.7 2.13 -99.2 2.53 -97.7 2.04
Z -114.0 1.80 -97.5 1.88 -97.2 1.80
===========================================================================
Potential energy maps also differed considerably. The quasi three-dimensional potential energy surface was table-like for the E isomer with ridges of very small heights between the shallow valley-like minima. There were sharp maxima when the hydrogens on the phenyl groups approached each other. The the map upside down). The two sets of almost symmetrical minima network were connected by relatively high energy hills, which corresponded to a near parallel arrangement of the phenyl groups.
Discussion
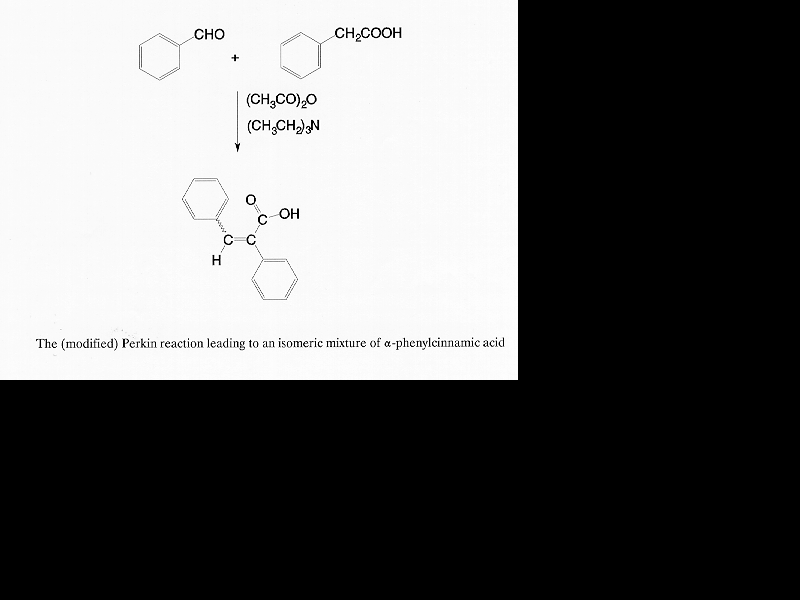
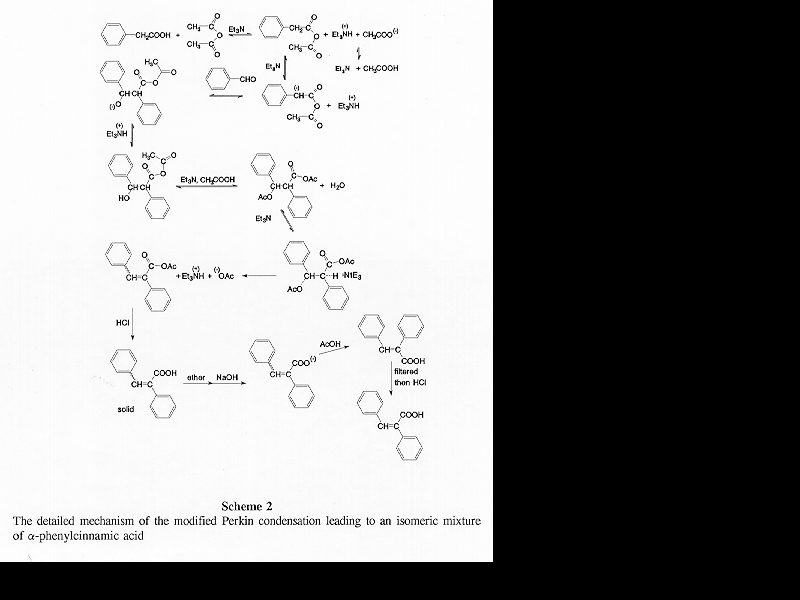
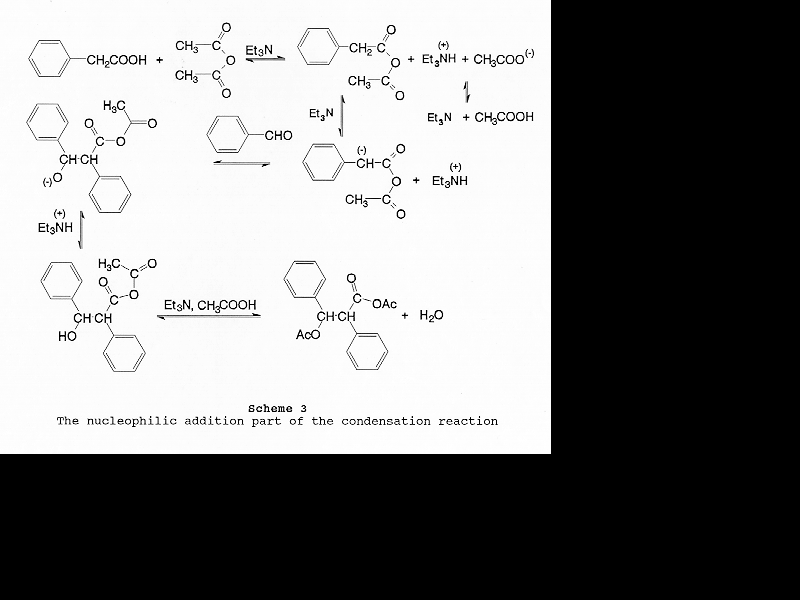
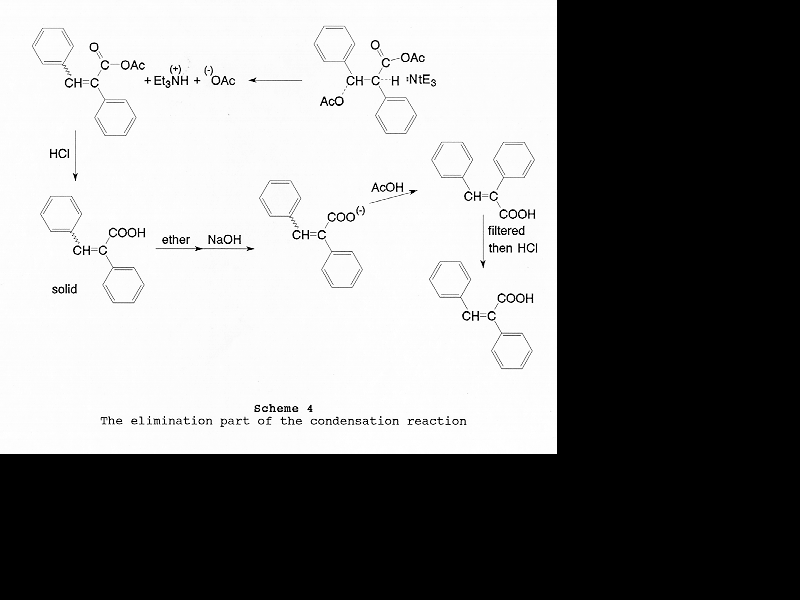
Based on published information [6, 15-17] and common sense, a detailed mechanism of this modified Perkin condensation can be put together (Scheme 2). It consists of two parts: (1) nucleophilic addition of the mixed anhydride formed from acetic anhydride and phenylacetic acid on the carbonyl group of benzaldehyde via base catalysis (Scheme 3); (2) the elimination of acetic acid and the hydrolysis of the product anhydride (Scheme 4). Zimmermann et al. have shown that the condensation reaction is elimination controlled [6]. There is no reason to assume that preferential formation of any of the four intermediates (Table 5) takes place.
{kind=link}
{kind=link}
{kind=link}
Table 5
The absolute configurations of the diastereomers and those of the products after elimination (for graphical view)
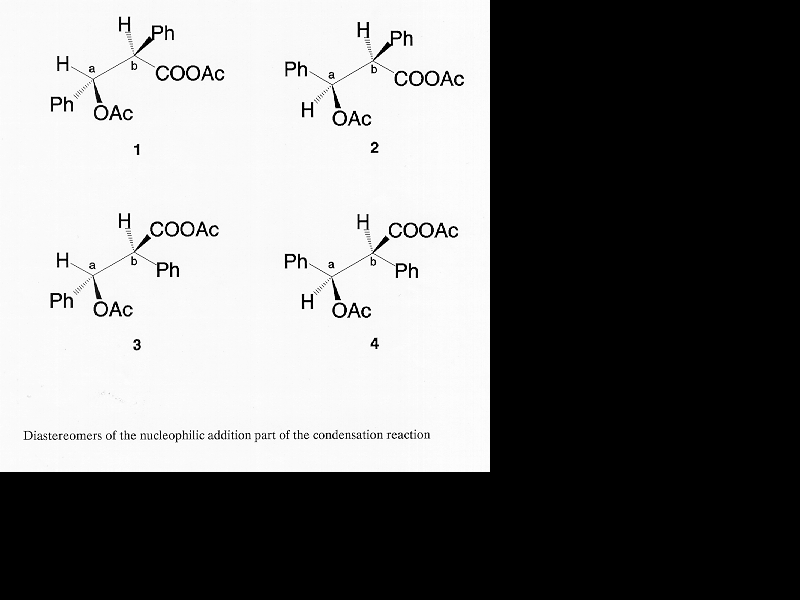{kind=link}
=================================================================================compounds chiral centers products after elimination ____________________________________
a b _________________________________________________________________________________
1 S R Z
2 R R E
3 S S E
4 R S Z
=================================================================================
According to AM1 semiempirical quantum chemical calculations, the heats of formation are very close to each other, their dipole moments are not too different either (Table 6). Also there is no need to assume other than the usual anti elimination [18], it can account for the formation of both isomeric acids.
Table 6
Standard heat of formation data (in kJ/mol) and dipole moments (D in Debye) for the isolated intermediates obtained by the AM1 method
=================================================================
compounds deltaHf,298 D
_________________________________________________________________
1 -657.8 5.48
2 -640.2 5.51
3 -652.8 5.24
4 -640.8 5.45
=================================================================
As concerns the stability data of the intermediates and the alpha-phenylcinnamic acid stereoisomers it is safe to state that the elimination reaction is endothermic. (It is strictly true for gas-phase reactions, nevertheless, it should be approximately valid in our system as well.) Thus, the transition state is product-like, therefore, the potential energy surfaces of the products can be used in explaining stereoselectivity towards the E isomer. As it has been already mentioned the potential energy surface for the E isomer contains extensive low-energy regions close to the absolute minimum throughout. The potential energy surface of the other isomers has several local minima but considerable amount of the surface is in the high energy region (saddles, ridges, local maxima). Consequently, the reason for the preferential formation of the E isomer is of kinetic origin, its formation is much faster than that of the Z. When reaction conditions are more severe this isomer also forms, but never in excess.
Conclusion
Using the modified Perkin condensation leading to alpha-phenylcinnamic acid isomers as model reaction, it has been demonstrated that experimental and computational methods together are able to give a more complete account of the observed stereochemical features than any of them could be alone.
Acknowledgement
This work was supported by the National Science Foundation of Hungary through grant F4297/1992. The financial help is highly appreciated.
References
[1] Mann, J., in Secondary Metabolism Ch. 4, p. 173, Clarendon Press, Oxford, 1987.
[2] Johnson, J., in Organic Reactions (Eds. Adams, R., Bachmann, W.F., Johnson, J.R., Fieser, L.F. and Snyder, H.R.) p. 210, John Wiley & Sons, Inc., Chapman & Hall, Ltd., New York and London, 1947.
[3] Buckles, R.E., J. Chem. Ed. 27 (1950) 210.
[4] Fieser, L.F., in Experiments in Organic Chemistry p. 182, Heath and Co., Boston, 1955.
[5] Buckles, R.E. and Bremer, K., in Org. Synt. Coll. Vol. 4, p.777, John Wiley & Sons, Inc., New York, London, Sydney, 1967.,
[6] Zimmermann, H. and Ahramjian, L., J. Am. Chem. Soc. 81 (1959) 2086.
[7] Dewar, M.J.S., Zoebisch, E.G., Healy, E.F. and Stewart, J.J.P., J. Am. Chem. Soc. 107 (1985) 3902.
[8] Dewar, M.J.S. and Thiel, W. J. Am. Chem. Soc. 99 (1977) 4899.
[9] Stewart, J.J.P., J. Comput. Chem. 10 (1989) 209, 221.
[10] Tasi, G., Pálinkó, I., Náray-Szabó, G., Halász, J., PcMolTM3.0, semiempirical Quantum Chemical Calculations on Microcomputers, Chemicro Ltd., Budapest, 1992.
[11] Stewart, J.J.P., QCPE Bull. 6 (1983) 43; MOPAC 6.0, QCPE program 455.
[12] Allinger, N.L., J. Am. Chem. Soc. 99 (1977) 8127.
[13] CacheTM, version 2.2, Tektronix Inc., licensed to Prof. Seddon, K.R., The Queen's University of Belfast, permission for use is highly appreciated.
[14] Török, B., Pálinkó, I., Tasi, Gy., Nyerges, L. and Bogár, F., J. Chrom. A 668 (1994) 353.
[15] Ogata, Y. and Tsuchida, M., J. Org. Chem. 24 (1959) 78.
[16] Ketcham, R. and Jambotkar, D., J. Org. Chem. 28 (1963) 1034.
[17] Buckles, R.E. and Cooper, J.A., J. Org. Chem. 30 (1965) 1588.
[18] Isaacs, N.S., in Physical Organic Chemistry p. 524, Longman Scientific & Technical, John Wiley & Sons, Inc., 1987; Carey, F. and Sundberg, R.J., in Advanced Organic Chemistry, Part A p. 377, Plenum Press, New York and London, 1993.