
No fluorine atom transfer can be detected by GC/MS analysis of the crude reaction mixture. Addition of a soluble fluoride source like tetrabutylammonium fluoride trihydrate, to a mixture of tetraphenyltin and 1-naphtyl iodide under the same conditions affords the coupled product with a comparable efficiency. Without added fluoride ion source no product can be detected under our conditions, which differ from the classical conditions of the Stille reaction mainly by the use of THF instead of HMPA as a solvent.
Addition of a carbon monoxide source like molybdenum hexacarbonyl leads to the formation of phenyl naphtyl ketone.
Palladium-catalyzed coupling of tetraorganotin compounds with aryl and benzyl halides[1], usually called " Stille reaction "[2] is a well documented reaction[3-6] with a wide number of applications, ranging from synthesis of natural products[7] to combinatorial chemistry[8].
One way to increase the scope of this reaction is to investigate new organotin species. For example internally coordinated 1-aza-5-stannabicyclo[3.3.3] undecane are by far more reactive than symmetrical tetralkyltin [9].
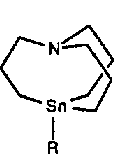
Those reagents efficiently transfer a methyl radical. Whereas pentacoordinated tin species are well known, there are few reports of their reactivity in organometallic chemistry. We wish to report here the reactivity of pentacoordinated tin species obtained by addition of fluoride to tetracoordinated tin compounds.
Tetrabutylammonium difluorotriphenylstannate 1 is a stable, crystallized compound useful as a synthetic equivalent of naked fluoride anion[10].
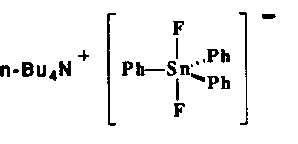
When 1 is reacted with 1-naphtyl iodide (2% Pd(PPh3)4, THF, reflux, 8h) the carbon-carbon coupled product expected from the Stille reaction, 1-phenylnaphtalene, was obtained in good yields (Table 1, entry 1).

No trace of fluorine atom transfer can be detected by GC/MS analysis of the crude reaction mixture. Under those conditions tetraphenyltin gave no coupling product (Table 1, entry 2), whereas fluorotriphenyltin led to a low yield.
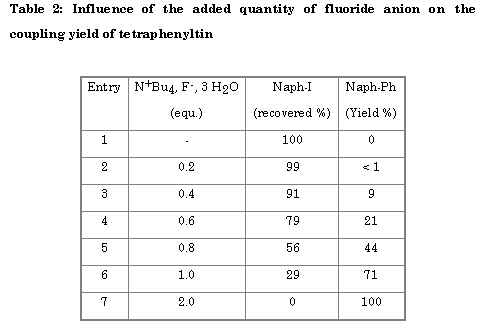
When fluoride ion was added to tetraphenyltin, yields increased (Table 2) and became nearly quantitative for two equivalents of tetrabutylammonium floride hydrate per tetraphenyltin.
Under those conditions, the reaction of fluoride ion with tetraphenyltin is fast, leading to the pentacoordinated fluoro tetraphenyltin anion. This in agreement with a reaction in which tin remains pentacoordinated:
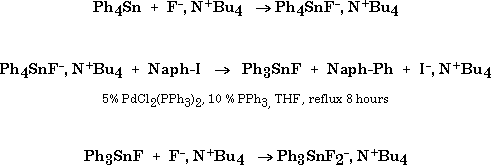
In presence of carbon monoxide the Stille reaction leads to the formation of ketone or aldahyde[11]. Molybdenum hexacarbonyl activated by fluoride on has been shown to be an efficient carbonylation system[12]. When molybdenum hexacarbonyl is added to our reaction mixture the expected naphtylphenylketone is obtained (Table 3).
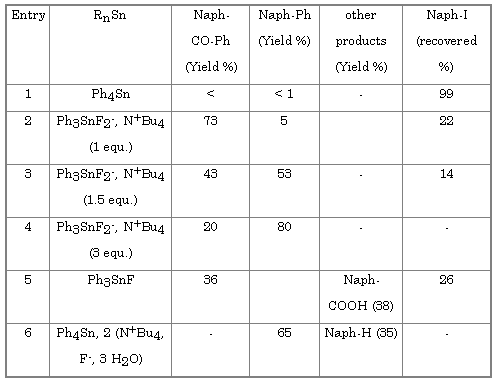
Surprisingly, the yields of ketone became lower when the number of equivalents of tetrabutylammonium difluorotriphenylstannate was increased (Table 3, entries 2 to 4). With 3 equivalents phenylnaphtalene is the major products. On the other hand the yield of ketone increases with the amount of molybdenum hexacarbonyl (Table 4).

Since early Stille's paper, LiCl has been shown to be an efficient catalyst of the coupling reaction of organotin and unsaturated iodide or triflate[13]. The role of the lithium has been explained by the formation of a more reactive catalyst involving a chlorinated palladium complex[14]. In a series of experiments including various salts K. Stille[13] pointed out that tetrabutylammonium fluoride was an efficient catalyst. However, contrary to lithium chloride tetrabutylammonium fluoride did not follow a second order kinetics, but the involvement of pentacoordinated tin has not been discussed.
This work shows that pentacoordinated tin are by far more reactive than the tetracoordinated species as was suggested by the reactivity of 1-aza-5-stannabicyclo[3.3.3][9]. The existence of a fluorinated complex of palladium and its role in the catalytic cycle are under investigation.
Pentacoordinated fluoro organotin species isolated or generated in situ from tetraaryl tin and tetrabutylammonium fluoride hydrate are efficient substrates in palladium-catalyzed coupling reaction with aromatic halide. The intimate mechanism of this reaction is under investigation.
[1] Kosugi, M; Sasazawa, K; Shimizu, Y.; Migita T. Chem. Letters 1977, 301
[2] Milstein, D.; Stille J. K. J. Am. Chem. Soc. 1979, 101, 4992-4998
[3] Stille, J. K. Angew. Chem. Int. Ed. Engl. 1986, 25, 508
[4] Tin in Organic Synthesis, Pereyre, M; Quintard, J.-P.; Rahm, A; Butterworths, London, 1987.
[5] Mitchell, T. N. Synthesis, 1992, 803
[6] Dawson, G. J.; Williams, J. M. J., Contemporary Organic Synthesis, 1994, 77
[7] Nicolaou, C.; Chakraborty, T. K.; Pisocopio, A. D.; Minowa, N.; Bertinato, P. J. Amer. Chem. Soc. 1993, 115, 4419
[8] Forman,F.; Sucholeiki, W. J. Org. Chem 1995, 60, 523-528
[9] Vedejs, E; Haight, A. R.; Moss, W. O., J. Amer. Chem. Soc. 1992, 114, 6556
10] Gingras, M. Tetrahedron Lett. 1991, 32, 7381-7384
[11] Tanaka, M, Tetrahedron Lett. 1979, 28, 2601
[12] Imbeaux, M; Mestdagh, H; Moughamir, K; Rolando, C J. C. S., Chem. Comm. 1992, 1678-1679.
[13] Scott, W. J.; Stille, J. K. J. Amer. Chem. Soc. 1986, 108, 3033
[14] Amatore, C.; Jutand, A; Suarez, A J. Amer. Chem. Soc. 1993, 115, 9531