Correspondence to thurstonde@sci1.sci.port.ac.uk
In order to increase cytotoxicity through the production of DNA cross-links, PBD-dimers have been recently synthesised, comprising of two PBD units joined through their A-rings[2]. Similarly, with the aim of producing a PBD monomer with DNA-crosslinking activity, Confalone and co-workers have synthesised[3] a PBD analogue with an epoxide group substituted at the C11a-position (1), although no DNA-binding data was reported. However, subsequent computer modelling studies in this laboratory suggested that C8 would be the preferred position for attachment of a second alkylating group, as attachment at the C11a-position could sterically hinder fit within the minor groove as well as interfere with nucleophilic attack of the guanine N2 at the C11-position. For these reasons, an epoxide-containing PBD (2) was designed in which a 2,3-epoxypropaneoxy group is attached to the C8-position of the PBD.
The main difficulty associated with PBD synthesis is the formation of the unstable N10-C11 carbinolamine-imine functionality, to afford PBDs in practical yields[4]. For the target molecule 2, this problem is further complicated by the requirement to include an equally labile epoxide group[5]. These problems were overcome by the design of a synthetic route based upon a method for PBD synthesis initially reported by Hurley and co-workers[6], but later modified by Fukuyama and co-workers[7], involving cleavage of N10-trifluoroacetyl- or N10-allyl carbamate-protected PBD-carbinolamines respectively. However, for the synthesis of 2, an amine protecting group was required that would be stable to epoxidation conditions but easily removed in the presence of the labile epoxide group.
It has been reported that the 9-fluorenylmethyloxycarbonyl (Fmoc) group can be used to protect amines in high yields (88%-98%)[8], followed by cleavage with tetrabutylammonium fluoride (TBAF) at room temperature[9]. Other reports suggest that the epoxide group is relatively stable to fluoride ion at this temperature5. For these reasons, the Fmoc group was utilized for the synthesis of the epoxide-PBD (2).
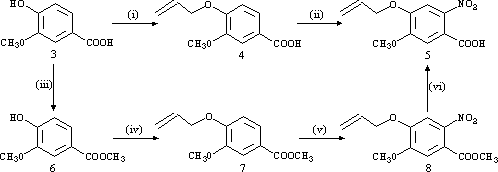
The propeneoxy-substituted A-ring (5) was first synthesised by two different routes using commercially available vanillic acid (3) as starting material. The first route involved conversion of 3 to its propeneoxy substituted analogue (4) with NaH/allyl bromide in THF, followed by nitration with SnCl4/HNO3 to give the desired A-ring (5). In the second route, 3 was esterified to its methyl ester (6) with H2SO4/CH3OH, and then substituted with the propeneoxy group using NaH/allyl bromide in DMF to give 7. The final two steps involved nitration with either SnCl4/HNO3 or 70% HNO3 to afford 8, followed by cleavage of the methyl ester using NaOH/THF. Although the second route used two additional steps, it resulted in a higher overall yield of 5 (73% compared to 55%) and a shorter overall reaction time.
Reagents: (i) THF/NaH/rt/1 h - THF/Allyl Bromide/0°C/45 min - D/5 days. (ii) CH2Cl2/SnCl4/HNO3/-25°C/10 min. (iii) CH3OH/H2SO4/D/24 h. (iv) DMF/NaH/rt/2 h - Allyl Bromide/rt/24 h. (v) Method A: CH2Cl2/ SnCl4/HNO3/-25°C/30 min. Method B: HNO3/0°C/1 h - rt/48 h. (vi) THF/NaOH(aq)/ rt/18 h.
(2S)-pyrrolidine-2-carboxaldehyde diethyl thioacetal (9), prepared via literature methods[4,10], was coupled to the propeneoxy-substituted A-ring (5) using DCC to afford the nitro thioacetal (10). The nitro group was reduced to the amine with SnCl2.2H2O in methanol to afford 11, followed by protection of the amine with Fmoc-Cl in dioxane to give 12 in high yield (88%). Cleavage of the diethyl thioacetal group with HgCl2/CaCO3 resulted in immediate ring closure to the Fmoc-protected carbinolamine (13). The final two steps of the synthesis involved epoxidation of the propeneoxy group with m-chloroperbenzoic acid to afford a diastereomeric mixture of the epoxide (14), followed by removal of the Fmoc protecting group with TBAF in DMF to form the N10-C11 imine while leaving the epoxide group intact. The resulting epoxide-PBD (2) was obtained in good yield (70%) and was fully characterised by NMR and MS[11].
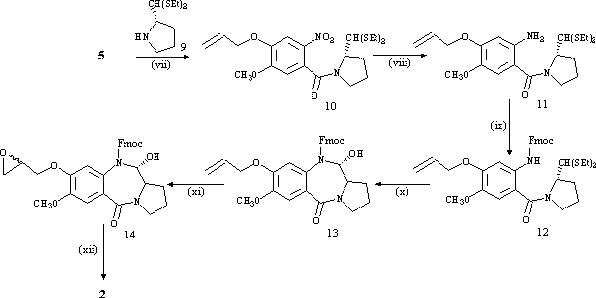
Reagents: (vii) CH2Cl2/DCC/rt/4 h - 9/rt/16 h. (viii) CH3OH/SnCl2.2H2O/D/3 h. (ix) Dioxane/Na2CO3(aq)/ Fmoc-Cl/0°C/4 h - rt/16 h. (x) CH3CN/H2O/HgCl2/CaCO3/rt/48 h. (xi) CH2Cl2/m-CPBA/rt/72 h. (xii) DMF/TBAF/rt/15 min.
The advantages of using Fmoc as a N10-protecting group for PBD synthesis, such as rapid cleavage under mild conditions, are apparent from this work. This strategy may be generally applicable to the synthesis of other PBD analogues, particularly those containing acid or base-sensitive functional groups. Preliminary studies indicate that 2 has significant DNA-binding properties and in vitro cytotoxicity, and this will be the subject of a future publication.
Acknowledgement
Financial support from the Cancer Research Campaign is gratefully acknowledged.
REFERENCES