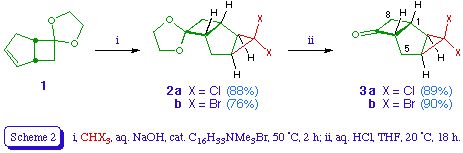
The starting point for this study was bicyclo[3.2.0]hept-2-en-6-one, which is available in two steps from cyclopentadiene [6]. We protected the ketone as the ethylene ketal 1 [7] to prevent any addition of carbene to the carbonyl group [8]. Treating 1 with dichloro- or dibromocarbene under phase transfer conditions [9] gave the respective tricyclic adducts 2a and 2b in good yield. Each adduct appeared to be a single stereoisomer, presumed to be derived from reaction on the more accessible exo face of the alkene. The adducts were readily transformed into the corresponding ketones 3a and 3b by hydrolysis with dilute hydrochloric acid in THF.
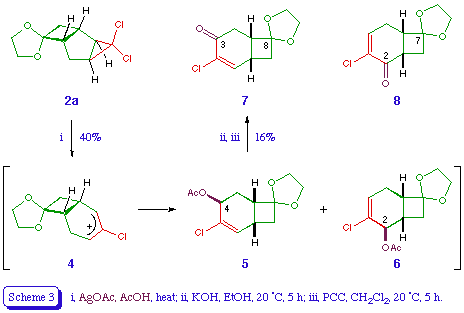
Ring expansion of the dioxolane 2a was attempted under various conditions. Thermolysis [10] at 200 for several hours gave a complex mixture of unidentified compounds. Heating with silver nitrate in aqueous acetone under reflux gave only starting material. However, treatment with silver acetate in acetic acid appeared to give the ring-expanded acetates 5 and 6 (total 40%, ratio 7:4), along with several unidentified compounds (Scheme 3). While the regiochemical relationship between the acetates 5 and 6 can be deduced by comparison of their n.m.r. spectra, the exo stereochemistry of the acetoxy group in each product is assigned on the basis of the reaction mechanism [5]. Hydrolysis and pyridinium chlorochromate (PCC) oxidation of the mixture of 5 and 6 afforded a low yield of a ketone, tentatively identified as 7 on the basis of the olefinic doublet in its proton n.m.r. spectrum (it is assumed that the corresponding signal in the regioisomer 8 would appear as a triplet). Similar complex mixtures and poor yields were observed on repeating the above sequence with the dibromocarbene adduct 2b.
The tricyclic ketones 3 proved more interesting from a synthetic viewpoint. Both reacted quite cleanly with silver acetate in acetic acid to give mixtures of ring expansion products, the best results being obtained with the dibromotricycle 3b (Scheme 4). Two stereoisomeric products were obtained in good yield, and easily distinguished by n.m.r. spectroscopy. The major product was 10 (50%), in which the olefinic (2-H) and carbinol (4-H) signals appeared as doublet and triplet respectively. In the minor regiosiomer 11 (25%) the olefinic signal (4-H) was more complex and the carbinol (2-H) signal narrower. A minor product 12 was identified as the cyclooctatrienone 12 (4%), which presumably arises via loss of a proton from the intermediate cation 9 (or a related species), leading to the formation of the bicyclo[4.2.0]octa-2,4-dienone 13, which undergoes valence tautomerisation to give the observed product [11].
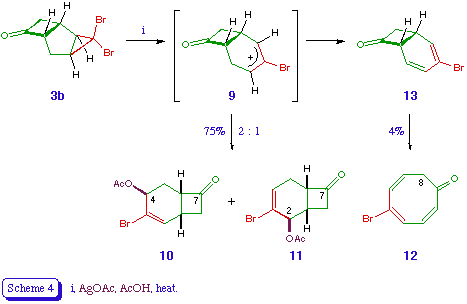
The exo location of the acetoxy groups in 10 and 11 is again assumed on the basis of the reaction mechanism, which involves the silver-assisted expulsion of bromide from 3b and the disrotatory cyclopropyl-allyl rearrangement to (effectively) an allyl cation 9 [5]. The orbital symmetry requirements of the process dictate that the bromine atom which is trans to the outward-rotating atoms must be expelled [12], and the mechanical constraints of the bicyclic system are such that only the hydrogen atoms at C-2 and C-4 can rotate outwards (Scheme 5). The nucleophile is thus delivered to the exo surface of the developing electrophile.
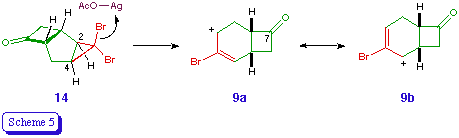
The preferential formation of regioisomers derived from the (formal) cation 9a rather than 9b is presumably a reflection of the greater stabilisation of its electrophilc centre, e.g. through hyperconjugation. A judicious choice of donor or acceptor substituent at C-2 or C-4 would thus appear to offer, through its electronic effect on the developing cationic centre, an obvious means of ensuring high and predictable regioselectivity in the ring expansion step [13].
EXPERIMENTAL
All compounds are racemic. M.p.s were determined on an Electrothermal apparatus and are uncorrected. Unless otherwise stated, i.r. spectra were of liquid paraffin mulls on sodium chloride plates, recorded on Perkin-Elmer 257 or 297 spectrometers. N.m.r. spectra were measured for solutions in deuteriochloroform unless otherwise indicated, with tetramethylsilane as the internal standard, on Varian EM 360 (1-H at 60 MHz), Varian CFT-20 (1-H at 80 MHz, 13-C at 20 MHz), or Perkin-Elmer R32 (1-H at 90 MHz) instruments. Mass spectra were measured on a Kratos MS30 instrument with a 70 eV electron impact source, and the peak abundances are quoted as a percentage of the base peak.
Starting materials and solvents were routinely purified by conventional techniques [14]. Distillation of liquid products was performed using a bulb-to-bulb (Kugelrohr) apparatus, and the temperatures quoted are those of the oven. Organic solutions were dried using anhydrous magnesium sulphate and concentrated by rotary evaporation. Analytical thin layer chromatography (t.l.c.) was carried out on Camlab Polygram SIL G/UV254 silica gel or ALOX N/UV254 alumina plates. Preparative (column) chromatography was carried out using Camag 100-250 mesh alumina, or 60H silica gel (Merck 7736) and hand-bellows pressure. Compositions of solvent mixtures are quoted as ratios of volume. 'Petroleum' refers to light petroleum, b.p. 40-60 'Ether' refers to diethyl ether.
spiro[Bicyclo[3.2.0]hept-2-en-6,2'-[1,3]dioxolane] 1 [7]
Bicyclo[3.2.0]hept-2-en-6-one (10.8 g, 0.10 mol), ethylene glycol (16 g, 0.26 mol), and Amberlystreg. 15 ion exchange resin (0.4 g) in dry benzene (100 ml) were heated under reflux in a Dean and Stark water trap for 24 h. The reaction mixture was then cooled, filtered, and the filtrate washed with saturated aqueous sodium bicarbonate (100 ml), water (3 x 100 ml), and brine (100 ml). Drying, followed by evaporation and distillation (91 17 mmHg) gave the title compound 1 (10.5 g, 69%) as a colourless oil; delta H (ppm) (60 MHz) 2.05 (1 H, dd, 7-Hendo), 2.3-2.8 (3 H, m, 4-H2 and 7-Hexo), 2.9-3.3 (2 H, m, 1-H and 5-H), 3.8 (4 H, s, 8-H2 and 9-H2), and 5.74 (2 H, s, 2-H and 3-H).
3,3-Dichlorospiro[tricyclo[4.2.0.02,4]octane-7,2'-[1,3]dioxolane] 2a
To a stirred mixture of spiro[bicyclo[3.2.0]hept-2-en-6,2'-[1,3]dioxolane] 1 (3.3 g, 21.7 mmol), chloroform (5.5 ml, 8.21 g, 69 mmol), and cetyltrimethylammonium bromide (58 mg, 0.16 mmol) at 50+/-2 was added dropwise over 15 min a solution of sodium hydroxide (6.75 g, 0.17 mol) in water (14 ml). The reaction mixture was stirred for further 2 h at the same temperature, and then treated with ice-water (30 ml), acidified with 10% sulphuric acid, and extracted with ether (3 x 30 ml). The ethereal layer was washed with water and brine, dried, and evaporated, and the residual oil distilled (110-112 1.5 mmHg) to obtain the title compound 2a (4.5 g, 88%) as a colourless oil (Found: C, 51.12; H, 5.11. C10H12Cl2O2 requires C, 51.09; H, 5.14%); delta H (ppm) (90 MHz) 1.9-2.8 (8 H, m) and 3.75-3.95 (4 H, m, 9-H2 and 10-H2); delta C (ppm) 105.5 (7-C), 67.2 (3-C), 64.0 (4'-C or 5'-C), 63.1 (4'-C or 5'-C), 53.8 (6-C), 42.4 (1-C), 38.7 (2-C or 4-C), 37.6 (8-C), 34.2 (2-C or 4-C), 28.1 (5-C); m/z 238 (M+, 37Cl2), 236 (M+, 37Cl, 35Cl), 234 (M+, 35Cl2), 199 (M-Cl), 164 (M-Cl2).
3,3-Dibromospiro[tricyclo[4.2.0.02,4]octane-7,2'-[1,3]dioxolane] 2b
The title compound 2b was prepared from 1 and bromoform as described for the dichloro analogue 2a. Distillation (102 0.1 mmHg) gave the title compound 2b (76%) as a pale yellow oil which later formed a white waxy solid, m.p. 35-45 delta H (ppm) (60 MHz) 1.9-3.4 (8 H, m) and 3.95-4.2 (4 H, br s, 9-H2 and 10-H2). The crude material was used without further purification for the preparation of the ketone 3b.
3,3-Dichlorotricyclo[4.2.0.02,4]octan-7-one 3a
A mixture of the dioxolane 2a (4.7 g, 20 mmol), hydrochloric acid (4 M; 40 ml), and tetrahydrofuran (40 ml) was stirred at room temperature overnight. The tetrahydrofuran was then evaporated under reduced pressure and the residue extracted with ether (3 x 30ml). The ethereal layer was washed with water, saturated aqueous sodium bicarbonate, and again water, dried, and evaporated. Distillation of the residue (59-60 0.1 mmHg) gave the title compound 3a (3.4 g, 89%) as a colourless oil (Found: C, 50.02; H, 4.16. C8H8Cl2O requires C, 50.29; H, 4.22%); vmax (neat) 1780 cm-1; delta H (ppm) (60 MHz) 2.1-2.5 (4 H, m, 2-H, 4-H, 5-H2), 2.6-3.7 (4 H, m, 1-H, 6-H, 8-H2); delta C (ppm) 209.4 (7-C), 67.25 (6-C), 66.4 (3-C), 49.9 (8-C), 42.2 (1-C), 38.4 (2-C or 4-C), 34.2 (2-C or 4-C), and 30.2 (5-C); m/z 194 (M+, 37Cl2), 192 (M+, 37Cl, 35Cl), 190 (M+, 35Cl2), 148 (M-C2H2O), 113 (M-C2H2OCl).
3,3-Dibromotricyclo[4.2.0.02,4]octan-7-one 3b
The title compound 3b was prepared from 2b (3.0 g, 9.26 mmol) as described for the dichloro analogue 3a. Crystallisation from petroleum afforded the title compound 3b (2.3 g, 89%) as colourless needles, m.p. 77-78 (Found: C, 34.41; H, 2.90. C8H8Br2O requires C, 34.32; H, 2.88%); vmax 1770 cm-1; delta H (ppm) (60 MHz) 1.8-2.4 (4 H, m, 2-H, 4-H, 5-H2), 2.5-3.6 (4 H, m, 1-H, 6-H, 8-H2); delta C (ppm) 209.2 (7-C), 67.5 (6-C), 49.7 (8-C), 43.4 (1-C), 39.55 (2-C or 4-C), 38.1 (3-C), 35.9 (2-C or 4-C), and 32.9 (5-C); m/z 282 (M+, 81Br2), 280 (M+, 81Br, 79Br), 278 (M+, 79Br2), 240, 238, 236, 198, 185, 183, 171, 169, 154, 152.
4-Chlorospiro[bicyclo[4.2.0]oct-4-ene-8,2'-[1,3]dioxolane]-3-one 7 [or the regioisomer 8]
A stirred mixture of silver acetate (0.85 g, 5.1 mmol) and anhydrous sodium acetate (2 g, 24 mmol) in Analar-grade acetic acid (20 ml) were heated under reflux for 20 min. The dioxolane 2a (1.175 g, 5 mmol) was then added and the stirring under reflux continued for a further 16 h. The hot mixture was then filtered, water (80 ml) was added, and the products extracted into ether (3 x 30ml). The extract was washed with water, saturated aqueous sodium bicarbonate, and again water, dried, and evaporated. Chromatography of the residue on silica gel, eluting with ether - petroleum (3:7) gave, along with various unidentified oils, an inseparable mixture of the title compounds 5 and 6 (total 0.52 g, 40%); delta H (ppm) (90 MHz) 2.05 (6 H, s, Me of 5 and 6), 1.8-2.9 (12 H, m, 1-H, 5-H2, 6-H, and 8-H2 of 5 and 6), 3.87 (8 H, br s, OCH2CH2O of 5 and 6), 5.22 (1 H, br s, 2-H of 6), 5.55 (1 H, t, J 5 Hz, 4-H of 5), 6.08 (1 H, dd, J 3.5 Hz, 2-H of 5), and 6.22 (1 H, dd, J 4.5, 5 Hz, 4-H of 6); m/z 260 (M+, 37Cl), 258 (M+, 35Cl), 223, 215, 201, 199, 163, 112, 106, 93, and 84. Compounds 5 and 6 were not separable by column chromatography. The 1H n.m.r. spectrum of the mixture showed a mixture of 5 and 6 in a ratio of 7:4. The noise-decoupled 13C n.m.r. spectrum of the mixture contained 19 peaks, indicating the presence of two major products.
A mixture of 5 and 6 (0.74 g, 2.86 mmol), and potassium hydroxide (0.25 g, 4.5 mmol) in ethanol (10 ml) was stirred at room temperature for 5 h. Water was then added, the ethanol was evaporated under reduced pressure, and the residue was acidified with hydrochloric acid. The mixture was then extracted with ether (3 x 25 ml), and the ethereal layer washed with water, dried, and evaporated. Chromatography of the residue on silica gel, eluting with ether - petroleum (1:1) gave the corresponding hydroxy compounds (0.5 g, 81%) as a colourless oil, which was stirred with pyridinium chlorochromate (0.75 g, 3.48 mmol) in dichloromethane (6 ml) at room temperature for 2.5 h. Ether was then added, the mixture was filtered through magnesium sulphate, and the ethereal filtrate was washed with water, dried, and evaporated. Chromatography of the residual brown solid (0.4 g) over alumina, eluting with ether - petroleum (3:7), followed by crystallisation from ether - petroleum, gave 4-chlorospiro[bicyclo[4.2.0]oct-4-ene-8,2'-[1,3]dioxolane]-3-one 7 [or the regioisomer 8] (0.1 g, 20%) as a colourless crystals, m.p. 98-99 (Found: C, 56.02; H, 5.18. C10H11ClO3 requires C, 55.96; H, 5.17%); vmax 1680 and 1605 cm-1; delta H (ppm) (60 MHz) 2.2 (1 H, d, J 11 Hz, 7-H), 2.5-3.4 (5 H, m, 1-H, 2-H2, 6-H, 7-H), 3.9 (4 H, br s, 4'-H2, 5'-H2), 7.0 (1 H, dd, J ca. 1, 3 Hz, 2-H).
4-Acetoxy-3-bromobicyclo[4.2.0]oct-2-en-7-one 10
2-Acetoxy-3-bromobicyclo[4.2.0]oct-3-en-7-one 11
5-Bromocycloocta-2,4,6-trien-1-one 12
The ketone 3b (5.6 g, 20 mmol) and silver acetate (3.34 g, 20 mmol) in acetic acid (100 ml) were heated under gentle reflux for 4 h. The hot reaction mixture was then filtered, washing the residue with hot water. A further portion of water (300 ml) was added to the filtrate, which was then extracted with ether. The ethereal layer was washed with water, saturated aqueous sodium bicarbonate, and again water, dried, and evaporated. The residual yellow oil was chromatographed over silica gel, eluting with ether - petroleum (1:1), which gave a mixture of the title compounds 10 and 11 (total 3.9 g, 75%) as a pale yellow oil, b.p. 112 (0.1 mmHg) (Found: C, 46.22; H, 4.35. C10H11BrO3 requires C, 46.36; H, 4.28%); vmax 1775 and 1725 cm-1; delta H (ppm) (90 MHz) [10] 1.75-2.0 (1 H, m, 5-H), 2.03 (3 H, s, COMe), 2.45 (1 H, overlapping ddd, J 2, 2.5, 15 Hz, 5-H), 2.75-3.7 (4 H, m, 1-H, 6-H, 8-H2), 5.45-5.55 (1 H, t, J 3 Hz, 4-H), 6.65 (1 H, d, J 5 Hz, 2-H); [11] 1.8-2.1 (1 H, m, 1-H), 2.1 (3 H, s, COMe), 2.2-2.6 (2 H, m, 5-H2), 2.7-3.7 (3 H, m, 6-H, 8-H2), 5.4-5.6 (1 H, m, 2-H), 6.45-6.55 (1 H, m, 4-H); m/z no M+; 216 (M-CH2CO), 179 (M-Br).
Another product isolated from the column was identified as the cyclooctatrienone 12 (0.16 g, 4%), which formed yellow crystals, m.p. 67 (petroleum) (Found: C, 48.15; H, 3.59. C8H7BrO requires C, 48.27; H, 3.54%); vmax 1645 and 1615 cm-1; delta H (ppm) (90 MHz) 3.17 (2 H, d, J 8 Hz, 8-H2), 5.75 (1 H, dd, J 8, 10 Hz, 7-H), 6.4 (1 H, d, J 10 Hz, 6-H), 6.5-6.9 (3 H, m, 2-H, 3-H, 4-H); delta C (ppm) 191.3 (1-C), 139.6 (5-C), 136.95, 135.4, 131.8, 130.35, 129.4, 43.4 (8-C); m/z 200 (M+, 81Br), 198 (M+, 79Br), 156 (M-CH2CO), 119 (M-Br).
ACKNOWLEDGEMENTS
We are grateful to the S.E.R.C. for financial support (Postdoctoral Fellowship GR/B/56470), and thank Roy Hayes and Ruth Howard for assistance with n.m.r. and mass spectroscopy.
REFERENCES
1 For a review of the natural occurrence and chemistry of cyclobutanones, see Bellus, D.; Ernst, B. Angew. Chem. Int. Ed. Engl. 1988, 27, 797.
2 For examples, see Grieco, P.A.; Oguri, T.; Wang, C.J.; Williams, E. J. Org. Chem. 1977, 42, 4113; Grieco, P.A.; Oguri, T.; Gilman, S. J. Am. Chem. Soc. 1980, 102, 5886; Smith, A.B.; Richmond, R.E. ibid. 1983, 105, 575; Wakamatsu, T.; Miyachi, N.; Ozaki, F.; Shibasaki, M.; Ban, Y. Tetrahedron Lett. 1988, 29, 3829.
3 For examples, see Parham, W.E.; Schweizer, E.E. Org. React. 1963, 13, 55; Jefford, C.W.; Burger, U.; Delay, F. Helv. Chim. Acta 1973, 56, 1083; Sadlo, H.; Kraus, W. Tetrahedron 1978, 34, 1965; Blanco, L.; Amice, P.; Conia, J.-M. Synthesis 1981, 289, 291.
4 Butt, S.; Davies, H.G.; Dawson, M.J.; Lawrence, G.C.; Leaver, J.; Roberts, S.M.; Turner, M.K.; Wakefield, B.J.; Wall, W.F.; Winders, J.A. Tetrahedron Lett. 1985, 26, 5077, and references cited therein.
5 For a mechanistic discussion, see Reese, C.B.; Shaw, A. J. Chem. Soc., Perkin Trans. I 1975, 2422.
6 Grieco, P.A. J. Org. Chem. 1972, 37, 2363.
7 Howard, C.C.; Newton, R.F.; Reynolds, D.P.; Wadsworth, A.H.; Kelly, D.R.; Roberts, S.M. J. Chem. Soc., Perkin Trans. I 1980, 852.
8 Greuter, H.; Winkler, T.; Bellus, D. Helv. Chim. Acta 1979, 62, 1275.
9 Joshi, G.C.; Singh, N.; Pande, L.M. Tetrahedron Lett. 1972, 1461.
10 For discussions and leading references, see Baird, M.S.; Lindsay, D.G.; Reese, C.B. J. Chem. Soc. (C) 1969, 1173; Jefford, C.W. Chimia 1970, 24, 357.
11 For the parent system, see Ganter, G.; Pokras, S.M.; Roberts, J.D. J. Am. Chem. Soc. 1966, 88, 4235.
12 Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry; Academic Press: New York, 1970; pp. 46-48.
13 For a pertinent example of this effect, see Baird, M.S.; Slowey, P.D. Tetrahedron Lett. 1982, 23, 3795.
14 Perrin, D.D.; Armarego, W.L.F.; Perrin, D.R Purification of Laboratory Chemicals; 2nd Edition, Pergamon: Oxford, 1980.