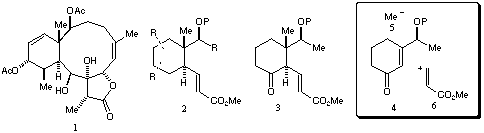
Scheme 1
Scheme 1
The approach chosen for our initial studies is outlined in the box shown in Scheme 1. Enone 4 would be subjected to conjugate addition of Me- (5) followed by trapping with a synthetic equivalent for synthon 6. This approach has been demonstrated to work for 3-methyl-2-cyclohexene-1-one using lithium dimethylcuprate as the nucleophile, and sulphoxide 7 as the synthetic equivalent for 6.[3] We prepared sulphoxide 7 on a large scale by modification of the literature procedure (Scheme 2).
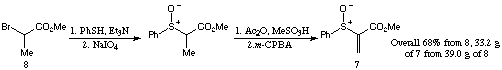 Scheme 2
Scheme 2Two substrates 9 and 10 were prepared in order to establish the effect of the protecting group on the diastereoselectivity of the initial conjugate addition. The preparation of these Michael acceptors was straightforward, the only noteworthy step being the chemoselective reduction of diketone 11 to 12 (Scheme 3).[4]
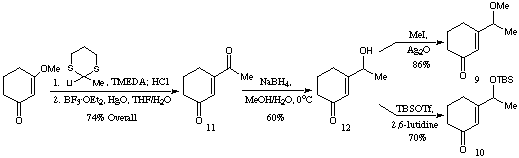 Scheme 3
Scheme 3Addition of lithium dimethylcuprate to the methyl ether 9 followed by attempts to trap the intermediate enolate with sulphoxide 7 in situ were unsuccessful, but this enolate could be trapped as the trimethylsilyl enol ether (Scheme 4). Treatment with acid gave ketone 13 as a 5:3 mixture of diastereoisomers. The crude silyl ether could also be used to generate a `clean' enolate which was be trapped relatively cleanly by sulphoxide 7. The product this reaction could be caused to undergo elimination to the desired enoate 14 (Scheme 4), which was clearly (proton nmr) a mixture of at least three diastereoisomers. In spite of the lack of stereochemical control this did demonstrate that the basic framework could be constructed using this approach, and that the overall yield (53% 9 -> 14) was acceptable, given the increase in molecular complexity.
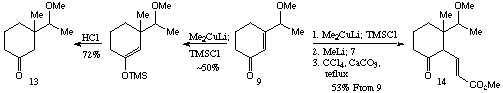 Scheme 4
Scheme 4With these results in hand, attention was turned to conjugate addition to the TBS protected enone 10. This would clearly be a much more suitable substrate as the hydroxyl protection is easily removed following subsequent transformations. Enone 10 was treated as above with lithium dimethylcuprate with a TMSCl trap. The diastereoselectivity of this conjugate addition was much improved over the corresponding O-methyl substrate, providing an excellent yield of a 7:1 mixture of diastereoisomers (Scheme 5). This could be deprotected in a stepwise manner to 15 and 16, and in an attempt to correlate these isomers with those obtained using the O-methyl substrate, alcohol 16 was treated with methyl iodide/silver oxide. The expected methyl ether was not isolated, the only product isolated being a volatile compound lacking a carbonyl group as a ~6:1 ratio of diastereoisomers. (Isolation was complicated by the volatility of 17 and it is possible that the slight change in diastereoisomer ratio is due to differing volatility of the isomers). This compound proved to be the bicyclic ketal 17. This ketal formation was fortunate, as it allowed determination of the stereochemistry using n.O.e. difference measurements, and showed that the major diastereoisomer from the conjugate addition was as shown in Scheme 5. The formation of the ketal was surprising as there is no evidence for appreciable amounts of the corresponding bicyclic hemiacetal in the proton nmr spectrum (vide infra).
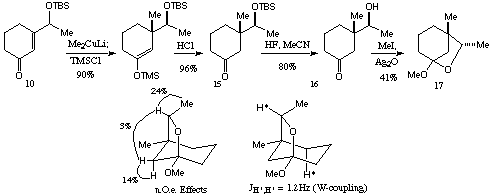 Scheme 5
Scheme 5There is a considerable body of work concerned with conjugate additions to unsaturated systems with a C-O bond adjacent to the site of attack, and several models have been proposed to account for the sense of diastereoselectivity observed.[5] The diastereoselectivity observed in our case is consistent with the `modified Felkin-Anh' model, in which the incipient sigma bond from the nucleophile is stabilized by interaction with the antibonding sigma* orbital of the C-O bond.[6] Whatever the details of the mechanism and stereochemistry, it is interesting to note that this reaction works as well as it does using lithium dimethylcuprate, as the closely related reaction shown in Scheme 6 gives very low yields in the absence a of Lewis acid.
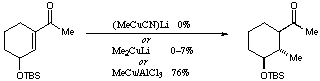
Scheme 6
Following the results described above, the final part of this initial investigation entailed trapping of the enolate from 10 with sulphoxide 7. This was carried out as before, to provide 15 in acceptable overall yield (~40%) from 10 as a 3:1 mixture of diastereoisomers after chromatography. This compound appeared to exist in the hemiacetal form as shown, and methylation gave the corresponding bicyclic ketal, the major diastereoisomer (16) of which could be isolated by careful chromatography. Proton nmr measurements confirmed the stereochemistry of this material to be as shown in Scheme 7.
Purification of intermediates on this route was difficult, and until hemiacetal 18 was reached, clean samples could not be obtained. In spite of this the overall yield from enone 10 is reasonable. In order to investigate future applications of this approach we also reacted the (crude) ketone 20 with LiAlH4 and MeLi. Although in both cases the yield was low (34% and 12%) the desired products 21 and 22 could be isolated, and proton nmr measurements supported the overall structural and stereochemical assignments.
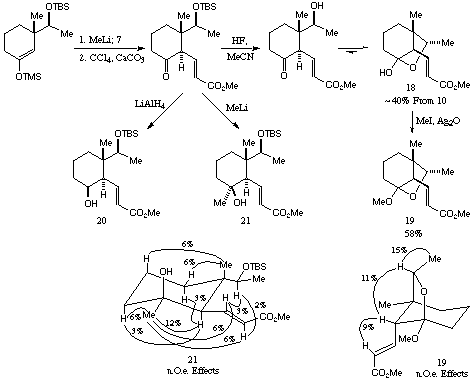 Scheme 7
Scheme 7A brief study of the feasibility of an enantioselective reduction of diketone 11 was made, as this would represent a simple method for achieving an enantioselective synthesis of our desired target. This asymmetric reduction would be interesting if successful, as the reaction would need to be regioselective as well as enantioselective. Although only two methods were tested,[7],8 the results were interesting, although inapplicable to the aims of this project. These experiments are illustrated in Scheme 8, in which the absolute configuration is assigned according to analogy with previous results for both these reagents with cyclohexenones.
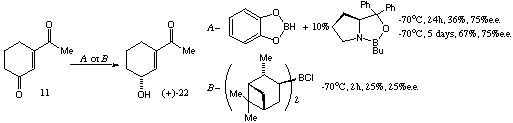 Scheme 8
Scheme 8It would appear from these preliminary experiments that, at least in principle, enantioselective reduction of a diketone such as 11 is possible in good enantiomeric excess (enantiomeric excesses estimated from proton nmr spectra in the presence of 3 equiv. of (R)-(-)-(9-anthryl)-2,2,2-trifluoroethanol).[9] Although the regioselectivity is unsuitable for the project described herein, the change of regioselectivity and the level of asymmetric induction are intriguing and warrant further investigation.
In conclusion, we have developed a relatively short route, which proceeds with reasonable stereochemical control, to intermediates with the potential for incorporation into a synthesis of the solenolides.
Acknowledgements We thank British Biotech for a studentship (M.B.R.), and Dr A.H. Davidson and Dr. M. Whittaker (British Biotech) for their interest in and contributions to this project.