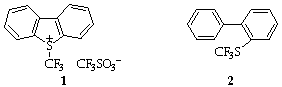
Recently, three methods for direct trifluoromethylation have come to the fore. 'CF3Cu' based organometallic methods,1 CF3SiMe3 (the Prakash reagent) based nucleophilic methods2 and S-trifluoromethyldibenzo[b,d]thiophenonium trifluoromethanesulfonate (TDTT) 1 based electrophilic methods.3 We here concentrate on the electrophilic reagent.
The TDTT reagent 1 and its analogues were introduced by Umemoto in 19903 and is particularly effective for the trifluoromethylation of a range of carbon and heteroatom nucleophiles. The reagent is stable, easily handled and is tunable by functionalisation of the benzene rings.4 The principle drawback lies in its long and inconvenient synthesis which uses gaseous CF3I for the trifluoromethylation step.5 As a prerequisite to further development of the reagent, we sought a convenient synthesis by which significant quantities would become available. We now report such a synthesis.
The initial target was the 2-fluoroalkylthiobiaryl 2. Biaryls are most conveniently made by one of the palladium catalysed cross coupling variants6 and incorporation of this process produced the synthetic scheme shown below.
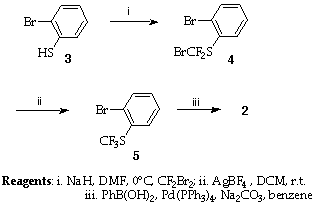
Scheme 1
Thus 2-bromothiophenol 3 was deprotonated and reacted with Burton's reagent (CF2Br2)5 under strictly anhydrous conditions, to give the bromodifluoromethyl thioether 4 (48%). This process is believed to occur by a carbene chain mechanism (Scheme 2)5 from which the susceptibility to protic quenching is apparent.
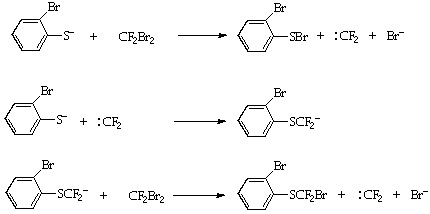
Scheme 2
A number of reagents were assessed for the fluorodebromination of 4, but best results (78%) were obtained with silver tetrafluoroborate in dichloromethane at room temperature.5 The product 5 was coupled under Suzuki conditions to give the initial target 2 (76%). Umemoto has reported a thorough study of the efficient conversion of 2 into the range of reagents of the type 14 and thus our synthesis of 2 provides a rapid, easily effected total route to these compounds.
Experimental
Melting points were carried out on a Kofler hot stage and are uncorrected; infrared spectra were recorded on Perkin Elmer 1700 FT spectrometer; 1H nmr spectra on a Bruker WH-250 FT (250 MHz), GE-300 FT (300 MHz), Jeol GSX FT (270 MHz-with a GSX data system) or Varian 60MHz spectrometer. Chemical shifts are reported in ppm relative to residual undeuteriated solvent, δ 7.26 for chloroform and δ 7.37 for benzene. 13C nmr spectra were recorded on the Bruker and Jeol instruments, with chemical shifts reported against CDCl3 reference.
Bromodifluoromethyl 2-bromophenyl thioether (4). -- Sodium hydride (0.164 g, 60% dispersion in mineral oil, washed [2 x 5 ml] with petroleum ether b.p. 40-60deg.C), was suspended in DMF (5 ml) and the stirred mixture treated at 0deg.C under nitrogen with a solution of 2-bromothiophenol (0.708 g, 3.74 mmol) in DMF (4 ml). After 15 min. at 0deg.C, dibromodifluoromethane (0.94 g, 0.409 ml) was added and the stirring continued for 1 h. The product mixture was diluted with ether (20 ml), washed with water (2 x 10 ml) and dried (MgSO4). The solvents were evaporated under reduced pressure and the residue purified by flash chromatography (SiO2; eluent: petroleum ether b.p. 40-60deg.C) to give bromodifluoromethyl 2-bromophenyl thioether (4) (0.57 g, 48%) as a colourless oil, νmax (Nujol)/cm-1 3040, 2900, 1570, 1545, 1440, 1410, 1108, 1060, 1015, 830, 745 and 640, δH (270 MHz, CDCl3/ppm) 7.87-7.78 (2H, m, Ar-H), 7.48-7.38 (2H, m, Ar-H); δF (90 MHz, CFCl3in CDCl3/ppm) -22.44; m/z 318 (M+), 239, 158, 108. Found: 319.8331, C7H4Br2F2S requires: 319.8328. Found: C, 27.14; H, 1.25; C7H4Br2F2S requires C, 26.44; H, 1.27%.
2-Bromophenyl trifluoromethyl thioether (5). -- Bromodifluoromethyl 2-bromophenyl thioether (4) (4.038 g, 12.7 mmol) in dichloromethane (10 ml) was added slowly at ambient temperature to a stirred suspension of silver tetrafluoroborate (2.719 g, 14.0 mmol) in dichloromethane (30 ml) under an atmosphere of nitrogen. After 15 min., the mixture was further diluted with dichloromethane (20 ml) and the insoluble material removed by filtration. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography (SiO2; eluent: hexane) to give 2-bromophenyl trifluoromethyl thioether (5) (2.508 g, 78%) as a colourless oil, νmax (Nujol)/cm-1 3050, 1570, 1560, 1455, 1430, 1130, 1100, 1025, 840, 755 and 650; δH (270 MHz, CDCl3/ppm) 7.86-7.76 (2H, m, Ar-H), 7.48-7.35 (2H, m, Ar-H); δF (90 MHz, CFCl3in CDCl3/ppm) -42.57 ppm; m/z 258 (M+), 189, 108, 69. Found: 257.9174; C7H4BrF3S requires 257.9148. Found: C, 32.69; H, 1.61; C7H4BrF3S requires C, 32.71; H, 1.57%.
2-Biphenyl trifluoromethyl thioether (2).3 -- 2-Bromophenyl trifluoromethyl thioether (5) (0.552 g, 2.15 mmol) in benzene (40 ml) was stirred under nitrogen with tetrakistriphenylphosphinepalladium (0.1 equiv.), aqueous sodium carbonate (8 ml of 1M solution) and phenylboronic acid (1 equiv.) in ethanol (5 ml) added. The mixture was refluxed under nitrogen until completion (tlc analysis) and the product extracted with ether (2 x 20 ml). The ethereal extract was washed with 10% aqueous disodium edetate (2 x 30 ml), water (3 x 30 ml) and dried (MgSO4). The solvent was removed under reduced pressure and the residue purified by flash chromatography (SiO2; eluent: petroleum ether 40-60deg.) to give 2-biphenyl trifluoromethyl thioether (2) (0.4152 g, 76%) as colourless crystals, m.p. 35-36deg.C; νmax (Nujol)/cm-1 3050, 3010, 1460, 1435, 1425, 1135, 1107, 1070, 755 and 700; δH (270 MHz, CDCl3/ppm) 7.90 (1H, d, J 7.6 Hz, H-3), 7.71-7.33 (8H, m, Ar-H); δF (90 MHz, CFCl3in CDCl3/ppm) -42.51 ppm; m/z 254 (M+), 185, 154, 69. Calc. for C13H9F3S, 254.0377; found: 254.0372. Calc. for C13H9F3S, C, 61.41; H, 3.57; found: 61.39; H, 3.61%.
2. I. Ruppert, K. Schlick and W. Volbach, Tetrahedron Lett., 1984, 25, 2195; G.K.S. Prakash, R. Krishnamurti and G.A. Olah, J. Am. Chem. Soc., 1989, 111, 393; D.R.Bellew, R. Krishnamurti and G.K.S. Prakash, J. Org. Chem., 1991, 56, 984.
3. T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579.
4. T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156.
5. M. Suda and C. Hino, Tetrahedron Lett., 1981, 22, 1997; I. Rico and C. Wakselman, Tetrahedron Lett., 1981, 22, 323.
6. A.Suzuki, N. Miyaura and T. Yanagi, Synthetic Commun. 1981, 11, 513; M.J. Sharp and V. Sniekus; Tetrahedron Lett., 1985, 26, 5997.