One of the most convincing methods for creating a new asymmetric center in a predictable configuration is the use of [3,3] sigmatropic rearrangements. The reason is that in such intramolecular rearrangements the favourable transition state geometry can normally be predicted from principles of conformational analysis3.
Encouraged by the high diastereoselective excess (de) obtained with (S)-proline ester in remote stereocontrol of thio-Claisen rearrangement4 using S-allylated derivatives of nitrothio-acetamides4b, several other chiral auxiliaries were screened for chiral induction studies.
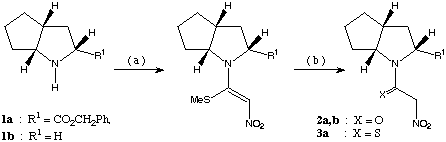
scheme 1: (a): 1,1-bis-methylthio-2-nitroethene, PTSA, MeCN, 12 h at rt, 6 h under reflux, (b): synthesis of 2a,b: HgCl2, MeCN : water ( 3 : 1 ), 12 h at rt, synthesis of 3a: Na2S and CH3CO2H in ethanol, 3 h at rt.
The bicyclic proline analogue 1a was chosen with the expectation of getting a higher de due to the steric bulk of the additional ring. In both cases the induction of chirality is caused by asymmetric centres located two atoms away from the C-carbon of the [3,3]-framework.
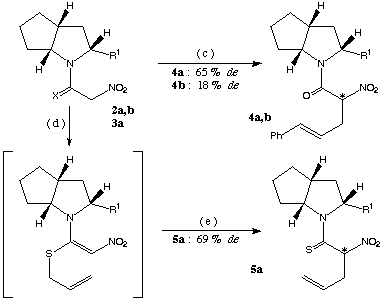
scheme 2: (c): 1. Pd(dba)2 (3 mol %), PPh3 (6 mol %) in acetonitrile, DBU (1 equiv.), cinnamyl acetate (1 equiv.), 8-15 h at rt, 2. 5 % HCl, at - 20[[ring]] C, (d): DBU (1.2 equiv.), allyl bromide in ethanol, (e): 2 h at rt.
The diastereoselectivity for the preparation of structure 5a (scheme 2) was only marginally better than that obtained with the (S)-proline ester4b used as chiral source (69 % de versus 66 % de).
Besides using substrates with nitrothioacetamides-structure, the chiral amino acid ester 1a was also tried as chiral auxiliary in our studies on the thio-Claisen rearrangement in other systems.
The diastereoselectivity obtained with benzoyl thioacetamide 6 (synthesis described in scheme 3) was much better than that obtained with the (S)-proline derivative4b (de: 33 %) forming the C-allyl derivative 7a with 57 % de.
The ester 1a was also converted to its nitroacetyl derivative (2a, synthesis shown in scheme 1) and subjected to Pd(0) catalyzed cinnamylation forming 4a. In this reaction also the chiral induction was far superior to the reaction with the corresponding (S)-proline ester4b (de: 45 %) leading to a diastereomeric excess of 65 %.
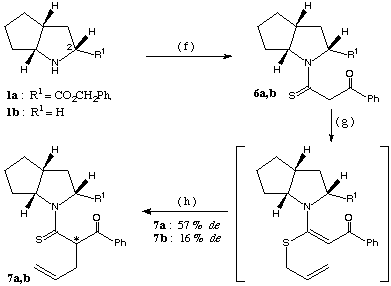
scheme 3: (f): methyl-benzoyldithioacetate, PTSA / MeCN, 8-10 h under reflux, (g): K2CO3 (1.2 equiv.) / acetone, allyl bromide (1.2 equiv.), 30 min. at RT, (h): CHCl3, 72 h at RT.
All the above reactions were repeated with the chiral bicyclic base 1b lacking the carboxylic ester at position 2 (scheme 3). The stereoselectivity was uniformly less than that reached with compound 1a as chiral auxiliary (de: 18 % for the preparation of product 4b and 16 % for the synthesis of compound 7b).
II. Stereoselective synthesis of optically active amines
In recent years, considerable attention has been devoted to the preparation of optically active compounds, mainly alcohols and amines. Different strategies provides access to enantiomerically pure amines: the diastereomeric addition of organometallic compounds to chiral ketimines5 or 1,3-oxazolidines6, the enantioselective catalytic hydrosilylation of ketoximes7, stereospecific transamination-alkylation-reactions8, diastereoselective α-alkylation of chiral modified amines9, the asymmetric hydroboration10 or the Grignard-addition to optically active nitrones11 .
This report describes the synthesis of chiral amines in presence of stoichiometric amounts of a chiral auxiliary 8.
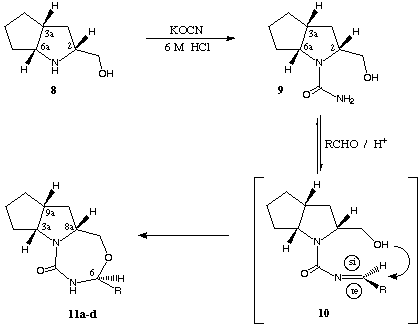
scheme 4
Starting from β-amino alcohol 8, e.g. the corresponding N-carbamoyl derivative 9, tri-cyclic compounds 11 (results in table 1) were synthesized with high stereoselectivity (diastereomeric ratio (dr) > 95 : 5, determination by 1H-NMR-spectroscopy using a sample of the crude product) via cyclisation with non-aromatic (R = i-propyl, c-hexyl) and aromatic aldehydes (R = phenyl, 1-naphtyl).
The cyclisation reaction is carried out under acid catalysis (with catalytic amounts of p-toluene sulfonic acid) in toluene with azeotropic removal of water (chemical yields between 57 and 86 %). Like the well established reaction of aldehydes with amino alcohols leading to 1,3-oxazolidines the condensation of aldehyd compounds with the NH2-function of 9 lead to the non-isolated imine-structures 10 (scheme 4). In the subsequent cyclisation-step the hydroxyl-function attacks the imine-intermediate stereospecific from the si-side (scheme 4 shows the favoured conformation of the postulated intermediate) transforming the former aldehyd carbonyl-function into a new stereogenic center (C-6 position, shown in scheme 4, change of numbers due to the change of the substitution pattern).
In order to ascertain unequivocally the absolute configuration at C-6 an X-ray crystal structure determination was performed on compound (3aR,6S,8aR,9aR)-11b. This compound exihibits 6S configuration confirming the proposed mode of addition, especially the stereochemical aspect of this reaction. Consequently nucleophilic attack occurs exclusively in the less sterically demanding situation as shown in scheme 4.
| compound | R | dra [%] | chem. yield [%]b | [α]S(20;D) |
|---|---|---|---|---|
| (3aR,6S,8aR,9aR)-11a | i-propyl | >= 95 : 5 | 57 | - 44.8 (1.81)c |
| (3aR,6S,8aR,9aR)-11b | c-hexyl | >= 95 : 5 | 73 | - 25.0 (1.04)c |
| (3aR,6S,8aR,9aR)-11c | phenyl | >= 95 : 5 | 77 | - 27.4 (0.99)c |
| (3aR,6S,8aR,9aR)-11d | 1-naphthyl | >= 95 : 5 | 86 | - 17.5 (1.00)d |
Diastereoselective ring-opening (scheme 5, again in all cases diastereomeric ratio > 95 : 5 determined by 1H-NMR analysis of the crude reaction mixture) leading to the formation of amides-structures 12 and subsequent reduction with the removal of the amino alcohol-unit 8 are the steps in achieving an asymmetric synthesis of optically active propylamines 13 (scheme 6):
The nitrogen bearing asymmetric center at C-6 is accessible for nucleophilic attack12 by organo metallic compounds furnishing the corresponding ring-opened products 12a,b,c and d. This reaction was carried out with a toluene solution of ZnEt2 (concentration: 15 %, excess of ZnEt2: 4.1 equiv.) under mild conditions (reaction temperature: 0[[ring]] C, yields between 89 and 91 %).
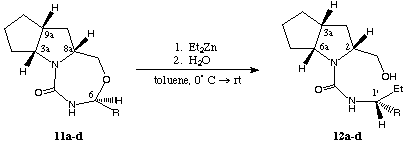
scheme 5
During the conversion with the organo metallic reactant the tricyclic compounds, respectively the directly involved stereogenic center undergo complete inversion of configuration (in all cases diastereomeric ratio (dr) > 95 : 5 determined by 1H-NMR analysis of the crude reaction mixture, results in table 2):
| compound | R | dr [%] | chem. yield [%] | [α]S(20;D) (c) |
|---|---|---|---|---|
| (all-R)-12a | i-propyl | >= 95 : 5 | 89 | - 30.1 (1.77)b |
| (all-R)-12b | c-hexyl | >= 95 : 5 | 91 | - 26.9 (1.30)b |
| (all-R)-12c | phenyl | >= 95 : 5 | 89 | - 35.0 (0.83)b |
| (all-R)-12d | 1- naphthyl | >= 95 : 5 | 89 | - 74.6 (0.98)c |
In the case of 12c,d one further transformation provides access to literature known chiral N-methyl-1-aryl-propyl amines obtained via reductive work-up with Red-Al (sodium bis[2-methoxyethyl]aluminium hydride).
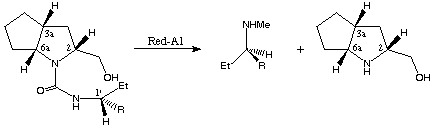
12c,d 13c,d 8
scheme 6
A comparison of the physical data, especially the optical rotation (table 3) demonstrates the high degree of diastereoselectivity for the nucleophilic attack and allows the determination of the obtained absolute configuration at C-1' (former C-6, scheme 5, change of numbers because of the change of the substitution pattern).
| compound | R | chem. yield. [%] | op [%] | [α]S(20;D) (c)a |
|---|---|---|---|---|
| (R)-13c | phenyl | 54 | 98 (R)b | + 36.1 (2.54) |
| (R)-13d | 1-naphthyl | 46 | 99 (R)c | + 53.9 (1.72) |
Both amines exhibit the R configuration, as well as both ring-opened starting compounds (1'R,2R,3aR,6aR)-12c,d, since reduction with Red-Al does not change the stereochemical properties of the resulting products.
In conclusion, the amino alcohol derivative of an industrial waste material 8, e.g. the corresponding N-carbamoyl derivative 9 represents a highly efficient source of chirality for the synthesis of optically active amines.
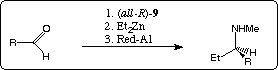
Because of internal asymmetric induction the chiral masked aldehydes (namely the tricyclic compounds (3aR,6S,8aR,9aR)-11) are diastereospecific transformed into optically active N-methyl-1-aryl-propylamines (op: 98 for 13c, respectively 99 % for 13d) in up to 37 % overall yield. Via the also available (all-S) enantiomers of 8 the corresponding S-configurated amines are also accessible
2 S. Wallbaum, T. Mehler, J. Martens, Synth. Commun. 1994, 24, 1381-1387.
3 a) E.R. Alexander and R.W. Kluiber, J. Am. Chem. Soc. 1951, 73, 4304. b) R.K. Hill and A.G. Edwards, Tetrahedron Lett. 1964, 3239. c) F.F. Ziegler, Acc. Chem. Res. 1977, 10, 227. d) G.B. Bennett, Synthesis 1977, 589. e) P.A. Bartlett, Tetrahedron 1980, 36, 3.
4 a) P. Belsin, S. Perrio, Tetrahedron 1992, 48, 4135. b) K.V. Reddy, S. Rajappa, Tetrahedron Lett. 1992, 33, 7957.
5 J. Yaozhong, L. Guilan, L. Jinchu, Z. Changyou, Synth. Commun. 1987, 17, 1545. 6 a) H. Takahashi, B.C. Hsieh, K. Higashiyama, Chem. Pharm. Bull. 1990, 38, 2429. b) A. Alberola, C. Andres, R. Pedrosa, Synlett 1990, 763.
7 H. Brunner, R. Becker, S. Gauder, Organometallics 1986, 5, 739.
8 A. Solladie-Cavallo, D. Farkhanj, Tetrahedron Lett. 1986, 27, 1331.
9 a) R.E. Gawley, K. Rein, S. Chemburkar, J. Org. Chem. 1989, 54, 3002. b) A.I. Meyers, D.B. Miller, F.H. White, J. Am. Chem. Soc. 1988, 110, 4778. c) I.M.P. Huber, D. Seebach, Helv. Chim. Acta 1987, 70, 1944.
10 H.C. Brown, K.-W. Kim, T.E. Cole, B. Singaram, J. Am. Chem. Soc.1986, 108, 6761.
11 Z.-Y. Chang, R.M. Coates, J. Org. Chem. 1990, 55, 3475.
12 a) H. Takahashi, I. Morimoto, K. Higashiyama, Chem. Pharm. Bull. 1990, 38, 2627. b) H. Takahashi, I. Morimoto, K. Higashiyama, Heterocycles 1990, 30, 287.