
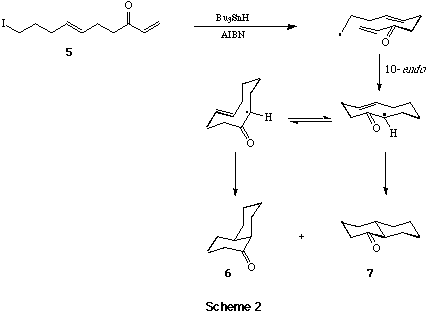
Cascade Macrocyclisation - Transannulation Reactions. - In some of our first investigations, we examined the radical macrocyclisation - transannulation sequence involving the iodotrienone 5, with a view to a 'one-pot' synthesis of 1-decalone.[11] Earlier work,[12] based on the precedent set by the studies of Porter et al,[13] had demonstrated the need for an electron deficient alkene electrophore, e.g. a conjugated enone, to promote macrocyclisations with nucleophilic radical centres. Thus, when a solution of 5 in benzene was heated in the presence of 1.1 equivs of Bu3SnH and a catalytic amount of AIBN for 0.5 h, workup and chromatography gave a 2:3 mixture of the cis-isomer 6 and trans-isomer 7 of 1-decalone, in a combined yield of 72%; treatment of this mixture with DBU (25°C, 24 hr) allowed the isolation of trans-1-decalone in essentially quantitative yield (Scheme 2).
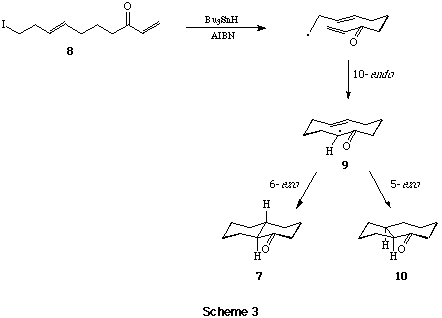
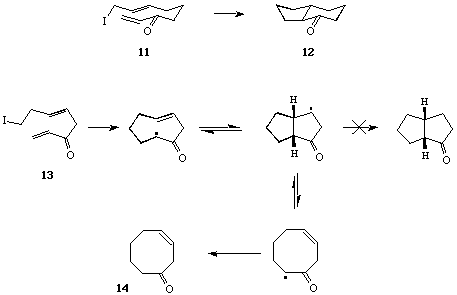
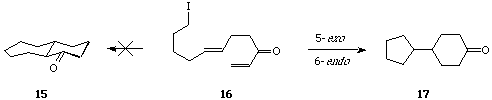
In further investigations of the scope for sequential radical macrocylisation - transannulations in bicycle constructions we showed that whereas the iodo-dienone 11 underwent tandem 9-endo-5-exo cyclisation producing the cis-tetralone 12 in reasonable yields (~50%), the corresponding E-octadienone 13 led only to the Z-cyclooctenone 14, and the iododienone 16 led to the 4-cyclopentyl substituted cyclohexanone (17; 95%), ie not to the anticipated 7,6-bicyclic ketone 15, on treatment with Bu3SnH-AIBN.
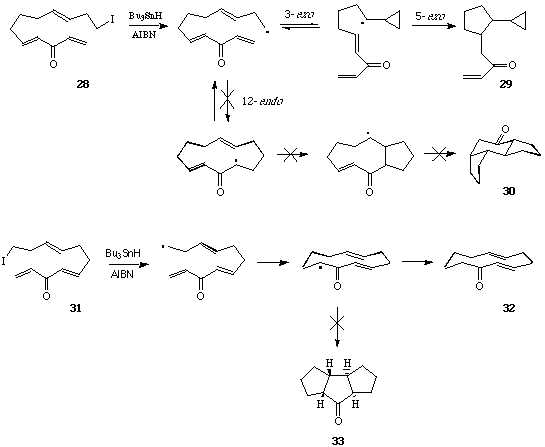
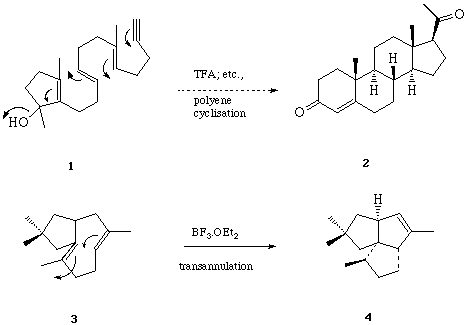
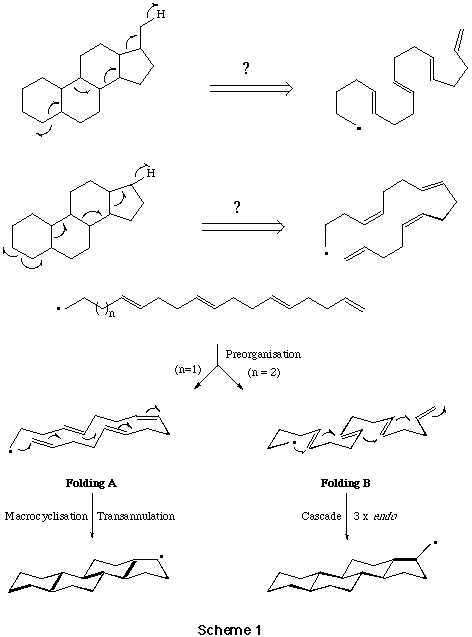
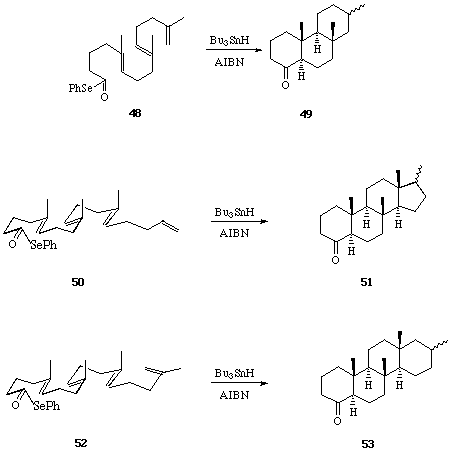
Sequential 6-Endo Trigonal Cyclisations. - It is now thirty years since Breslow et al,[19] and later Julia et al[20] first examined the possibility of a free-radical mechanism for the oxidative cyclisation of squalene, and more recently this hypothesis has been re-visited by Snider et al[21] and by Zoretic et al[22] amongst others.[23] The construction of fused polycycles by way of sequential radical mediated cyclisation reactions from alkyl centred radicals is well documented. Furthermore, with few exceptions 5-exo trig cyclisations are generally preferred over 6-endo trig closures from hex-5-en-1-yl radical intermediates,[24] and attempts to use consecutive 6-endo trig cyclisations from (5-, 9-, 13-) polyolefinic alkyl radical precursors in the formation of linear and angular 6-ring fused constructions have met with failure.[25] The unusual tendency of hex-5-en-oyl, i.e. acyl, radicals to cyclise via the 6-endo trig mode,[26] leading to six-ring carbocycles, has prompted us to evaluate the consecutive cyclisations of a range of (5-, 9-, 13-, 17-) polyolefinic acyl radical intermediates with a view to the synthesis of linear and angular fused 6-ring systems include steroid constructions.[27] We chose phenylselenyl esters as the most practical and convenient source of acyl radical intermediates.
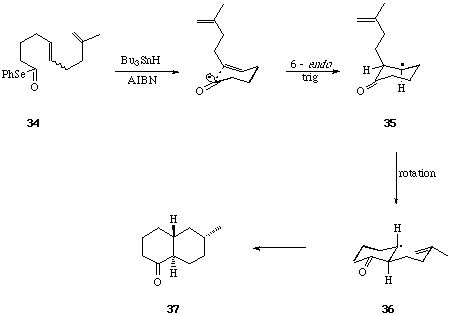
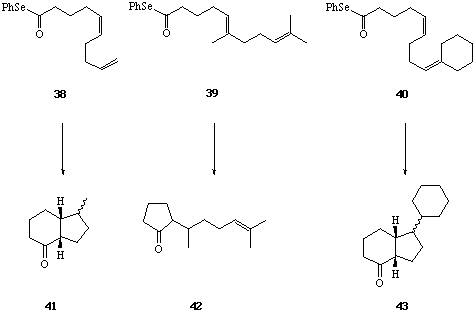
We first examined the cyclisations of the acyl radical intermediates produced from the Z- and E-isomers of the diene selenyl ester 34.[28] When solutions of the pure Z- and E-isomers of 34 were treated separately with Bu3SnH-AIBN (reflux 8 h), each was found to undergo two consecutive 6-endo trig cyclisations leading to the trans-decalone 37 in 70-80% yield. The formation of a single diastereoisomer of a single regioisomer of 37 from either the Z- or E-isomer of 34 is significant, and is best rationalised on the basis of: i, rapid inversion of the stereochemistry of the beta-keto radical intermediate (35 to 36) prior to the second ring forming reaction, and ii, preference for formation of the most stable radical product in the second 6-endo cyclisation leading to 37 from 36. The importance of substitution on the 5- and the 9- double bonds in 34 in determining the regiochemical outcome of the bi-cyclisation leading to 37, was demonstrated by cyclisations of the related phenyl selenyl esters 38, 39 and 40, to 41, 42 and 43 respectively.
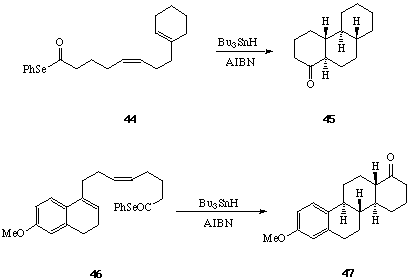
[1] For recent reviews see: J.K. Sutherland "Polyene Cyclisations", in Comprehensive Organic Synthesis, Vol 3, 341, Ed. B.W. Trost, Pergamon Press, 1991; S.K. Taylor, Org. Prep. Proc. Int., 1992, 24, 247.
[2] e.g. N.E. Carpenter, D.J. Kucera and L.E. Overman, J. Org. Chem., 1989, 54, 5846; A. de Meijere, F.E. Meyer and P.J. Parsons, J. Org. Chem., 1991, 56, 6487; M.J. Dorrity, R. Grigg, J.F. Malone, V. Sridharan and S. Sukirthalingham, Tetrahedron Lett., 1990, 31, 1343; Y. Shi and B.M. Trost, J. Am. Chem. Soc., 1992, 114, 791.
[3] e.g. P. Deslonghcamps, Aldrichim Acta, 1991, 24, 43.
[4] For bibliography see: W.B. Motherwell and D. Crich, Free Radical Chain Reactions in Organic Synthesis, Academic Press, London, 1991; C.P. Jasperse, D.P. Curran and T.L. Fevig, Chem. Rev., 1991, 91, 1237.
[5] N.J.G. Cox, G. Pattenden and S.D. Mills, Tetrahedron Lett., 1989, 30, 621; N.J.G. Cox, S.D. Mills and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1992, 1313; S.A. Hitchcock and G. Pattenden, Tetrahedron Lett., 1990, 31, 3641; S.A. Hitchcock and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1992, 1323.
[6] See for example: A.Ali, D.C. Harrowven and G. Pattenden, Tetrahedron Lett., 1992, 33, 2851; G. Pattenden and S.J. Reynolds, J. Chem. Soc., Perkin Trans. 1, 1994, 379.
[7] G. Pattenden, A.J. Smithies and D. S. Walter, Tetrahedron Lett, 1994, 35, 2413.
[8] G.J. Hollingworth, G. Pattenden and D. J. Schulz, Aust. J. Chem., 1995, 48, 381.
[9] For recent reference and bibliography see: P.V. Fish, A.R. Sudhakar and W.S. Johnson, Tetrahedron Lett., 1993, 34, 7849.
[10] G. Pattenden and S.J. Teague, Tetrahedron Lett., 1984, 25, 3021; Tetrahedron, 1987, 43, 5637.
[11] For some preliminary results see reference 7.
[12] For some summary of earlier work see: G. Pattenden, 'Polycycle Constructions by Transition Metal Catalysed and Radical Mediated Processes' in Organometallic Reagents in Organic Synthesis, Academic Press, eds. J.H. Bateson and M.B. Mitchell, 1993.
[13] See: N.A. Porter, D.R. Magnin and B.T. Wright, J. Am. Chem. Soc., 1986, 108, 2787; N.A. Porter, V.H.-T. Chang, D.R. Magnin and B.T. Wright, J. Am. Chem. Soc., 1988, 110, 3554; N.A. Porter, B. Lacher, V.H.-T. Chang and D.R. Magnin, J. Am. Chem. Soc., 1989, 111, 8309.
[14] S.A. Hitchcock and G. Pattenden, Tetrahedron Lett., 1992, 33, 4843 (corrigendum Tetrahedron Lett., 1992, 33, 7448).
[15] D.W. Pryde, Unpublished work; Nottingham University.
[16] For some preliminary results see: M.J Begley, G. Pattenden, A.J. Smithies and D.S. Walter, Tetrahedron Lett., 1994, 35, 2417.
[17] For an example of a radical-mediated transannular strategy towards diterpenoid ring systems see: A.G. Myers, K.R. Condronski, J. Am. Chem. Soc., 1993, 115, 7926; idem, ibid, 1995, 117, 3057.
[18] A.L.J. Beckwith and C.H. Schiesser, Tetrahedron, 1985, 41, 3925; K.N. Houk and D.C. Spellmeyer, J. Org. Chem., 1987, 52, 959; K.N. Houk and J.L. Broeker, J. Org. Chem., 1991, 56, 3651.
[19] R. Breslow, E. Barrett and E. Mohacsi, Tetrahedron Lett., 1962, 1207; R. Breslow, S.S. Olin and J.T. Groves, Tetrahedron Lett., 1968, 1837.
[20] M. Julia, Tetrahedron Lett., 1973, 4464.
[21] M.A. Dombroski, S.A. Kates and B.B. Snider, J. Am. Chem. Soc., 1990, 112, 2759.
[22] P.A, Zoretic, X. Wang and M.L. Caspar, Tetrahedron Lett., 1991, 32, 4819; P.A. Zoretic, Z. Shen, M. Wang and A.A. Ribeiro, Tetrahedron Lett., 1995, 36, 2925; P.A. Zoretic, Y. Zhang, and A.A. Ribeiro, Tetrahedron Lett., 1995, 36, 2929.
[23] e.g. M. Hoffman, Y. Gao, B. Pandey, S. Klinge, K.D. Warzecha, C. Kruger, H.D. Roth and M. Demuth, J. Am. Chem. Soc., 1993, 115, 10358.
[24] D.C. Spellmeyer and K.N. Houk, J. Org. Chem., 1987, 52, 959; A.L.J. Beckwith, Tetrahedron, 1981, 37, 3073; A.L.J. Beckwith and C.H. Schiesser, Tetrahedron, 1985, 41, 3925; D.P. Curran, Synthesis, 1988, 417; D.P. Curran, Synthesis, 1988, 489; M. Julia, Acc. Chem. Res., 1971, 4, 386.
[25] cf. E.R. Lee, I. Lakomy, P. Bigler and R. Scheffold, Helv. Chim. Acta., 1991, 74, 146.
[26] D.J. Coveney, V.F. Patel, G. Pattenden and D.M. Thompson, J. Chem. Soc., Perkin Trans. 1, 1990, 2721; T.M. Patrick, Jr., J. Org. Chem., 1952, 17, 1009, 1269; R. Dulou, Y. Chretien-Bessiere and H. Desalbres, C.R. Acad. Sci., Ser. C, 1964, 258, 603; J-P. Montheard, ibid., 1965, 260, 577; M. Chatzopoulos and J-P. Montheard, ibid., 1975, 280, 29; J.A. Kampmeier, S.H. Harris and D.K. Wedegaertner, J. Org. Chem., 1980, 45, 315; M. Julia and M. Maumy, Bull. Soc. Chim. Fr., 1969, 2415; Z. Cekovic, Tetrahedron Lett., 1972, 749; E.J. Walsh, Jr., J.M. Messinger II, D.A. Grudoski and C.A. Allchin, Tetrahedron Lett., 1980, 21, 4409; P. Delduc, C. Tailham and S.Z. Zard, J. Chem. Soc., Chem. Comm., 1988, 308; D.L. Boger and R.J. Mathvink, J. Org. Chem., 1988, 53, 3377; D.L. Boger and R.J. Mathvink, J. Org. Chem., 1989, 54, 1779; D.L. Boger and R.J. Mathvink, J. Am. Chem. Soc., 1990, 112, 4003; D.L. Boger and R.J. Mathvink, J. Am. Chem. Soc., 1990, 112, 4008; D.L. Boger and R.J. Mathvink, J. Org. Chem., 1990, 55, 5442; D.L. Boger and R.J. Mathvink, J. Org. Chem., 1992, 57, 1429; D. Crich and S.M. Fortt, Tetrahedron Lett., 1987, 28, 2895; D.Crich and S.M. Fortt, Tetrahedron Lett., 1988, 29, 2585; D. Crich and S.M. Fortt, Tetrahedron, 1989, 45, 6581; D. Crich, K.A. Eustace and T.J. Ritchie, Heterocycles, 1989, 28, 67; D. Batty, D. Crich and S.M. Fortt, J. Chem. Soc., Chem. Commun., 1989, 1366; f) D. Batty, D. Crich and S.M. Fortt, J. Chem. Soc., Perkin Trans., 1, 1990, 2875; D. Crich, K.A. Eustace, S.M. Fortt and T.J. Ritchie, Tetrahedron, 1990, 46, 2135; D. Batty, D. Crich, Tetrahedron Lett., 1992, 33, 875.
[27] Both Boger et al and Crich et al have also examined aspects of tandem cyclisations of acyl radicals produced from certain unsaturated selenyl esters. See under reference 26.
[28] For preliminary results see: a) L. Chen, G.B. Gill and G. Pattenden, Tetrahedron Lett., 1994, 35, 2593. b) H. Simonian, Unpublished work; Nottingham University.