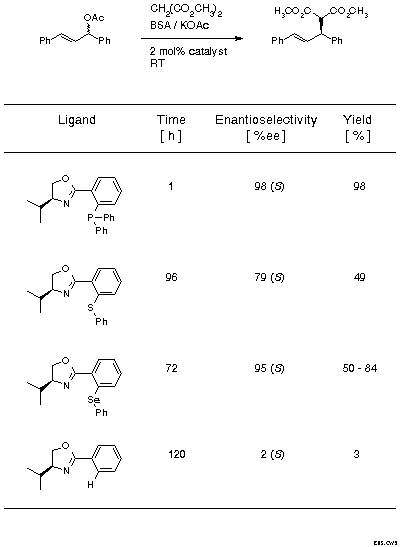
In the early phase of exploring ligands with two different donor atoms it was important to establish the best combination of donor atoms, i.e., to evaluate which of the soft donor atoms, P, S, or Se, would be the best choice, given the nitrogen of the oxazoline which is indispensable as carrier of the chiral information. For this evaluation the standard test system in the area, the reaction of 1,3-diphenylallyl acetate with dimethyl malonate, was chosen (Scheme 8) and ligands derived from valine were employed. In addition to the P, S, and Se derivatives, the monodentate phenyl oxazoline was also tested. The reaction times and yields indicate that the combination of N with P is by far the most effective one. The monodentate ligand is inactive. Typically, the most reactive system is also the most selective. It is also significant that with all ligands the product with (S)-configuration is preferentially formed.
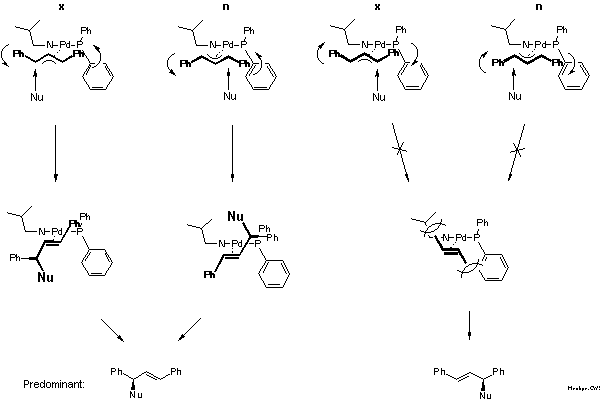
How can one rationalize the preferred steric course of the reaction: (S)-configuration? Answering this question for asymmetric ligands is much more difficult than for C2-symmetric ligands as is illustrated by Scheme 9. In the case of the pi-allyl-complex of the C2-symmetric bisoxazoline the nucleophile can attack either allylic terminus. The experimental decision between these possibilities is given by the configuration of the preferred product. As already explained in the introduction (cf. Scheme 4), preferential attack occurs at the terminus with R' and R in cis disposition. This is plausible because the adjacent C-Pd bond is the longer, and, therefore, the weaker bond.
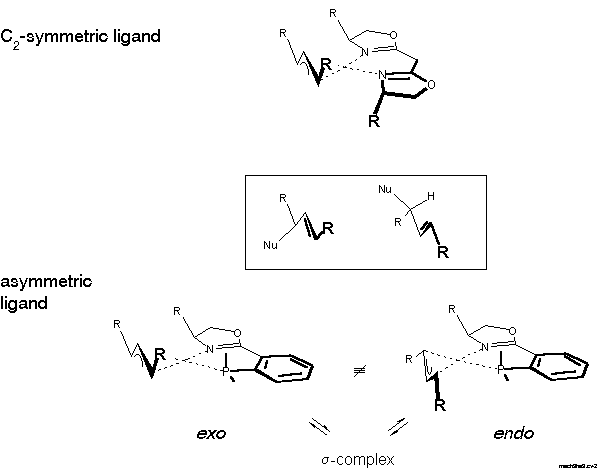
With an asymmetric ligand the problem is more difficult, because according to NMR there are two diastereomeric pi-allyl complexes, designated exo- and endo-isomer here. Interconversion of these isomers proceeds via sigma-allyl-complexes, by pi-sigma-pi interconversion[20], rather than simple rotation. The products can be formed via four pathways and the preferred product, again the enantiomer on the left-hand side of the box in Scheme 9, can arise by reaction at the C trans to P of the exo or cis to P at the endo isomer.
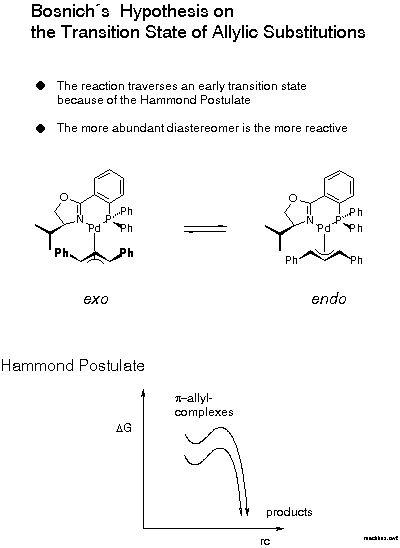
For the decision between these possibilities, a postulate put forward in 1985 by Bosnich[21] was helpful: the reaction traverses an early transition state. Bosnich argued that the energy content of a pi-allyl-complex is higher than that of the product and, therefore, that the transition state should be similar to the pi-allyl-complex, as stated by the Hammond postulate. Furthermore, in case of a fast equilibrium between intermediates, the reaction mainly proceeds via the transition state of lower energy according to the Curtin-Hammett principle. It follows that the more abundant isomer is the more reactive. Which of these isomers is the more stable? We were able to solve this problem [22]. Remarkably, before that there were predictions, even in print, that it would be the endo-isomer. The experimental determination first required preparation of the pi-allyl complexes.
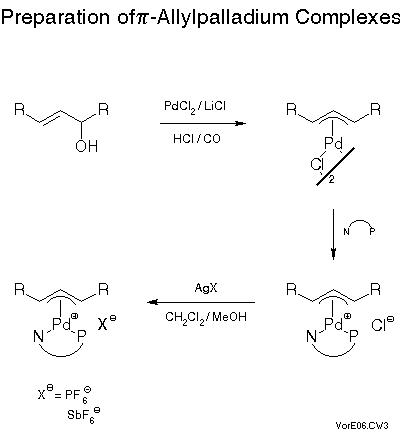
We were able to prepare a variety of pi-allyl complexes of the phosphinoxazolines via established methods[23] (cf. Scheme 11).The standard method involves first reaction of an allylic alcohol with lithium tetrachloropalladate under acidic and reducing - with carbon monoxide - conditions to give the air stable chloropalladium pi-allyl complex which simply is treated with the chiral ligand to give the chloro or chloride complex which is usually transformed, by treatment with a silver salt, into the hexafluorophosphate or hexafluoroantimonate. Salts of these anions often yield crystals suitable for x-ray analysis. The results were quite remarkable (Scheme 12).
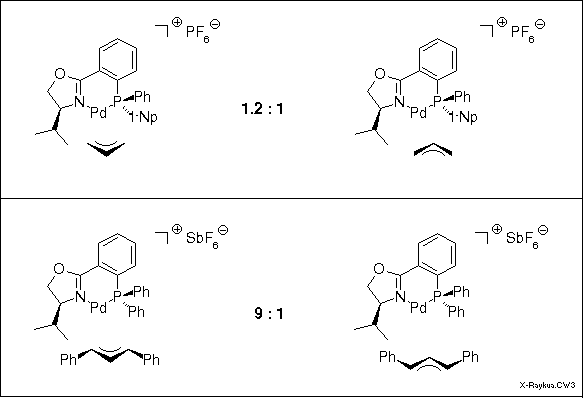
The crystal of the complex of the unsubstituted allyl system contained both the exo and the endo complex, only distinguished by the location of the carbon No 2. In CDCl3 solution NMR revealed a ca. 1:1 ratio. The diphenylallyl complex in solution displayed a 9:1 ratio of isomers, but surprisingly, it was the exo isomer that was found in the crystal. This was also the predominant isomer in solution as could be easily determined by dissolving a crystal at -78 deg C. At this temperature no isomerization occurs. Warming up to r.t. produced the 9:1 equilibrium ratio.
In conjunction with Bosnich's argument, lower activation energy for the more stable, now known to be the exo isomer, we infer from the known configuration of the product of allylic substitution that the nucleophile preferentially attacks the carbon trans to P (Scheme 13).
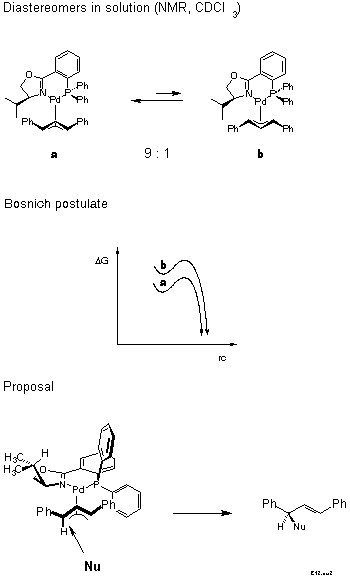
We have tried hard to directly verify this hypothesis by treating in a NMR tube the isomer mixture under non-equilibrating conditions with nucleophiles. Unfortunately, all nucleophiles catalyzed equilibration to occur much faster than substitution.
However, our hypothesis is further strengthened by the observation that according to 13C NMR the positive charge on the C trans to P is higher than that trans to N. Furthermore, as previously pointed out, interconversion of exo and endo diastereomers proceeds via the sigma-complex. We have closely studied these interconversions by NMR [22]. For the case of the unsubstituted allylic system it was found that the Pd-C bond trans to P, not trans to N, is opened to give the sigma-complex displayed in Scheme 14. From the sigma-complex one can obtain the endo-sigma-complex either by rotation around the Pd-C or the C-C-bond. Exchange experiments show that only C-C rotation occurs.
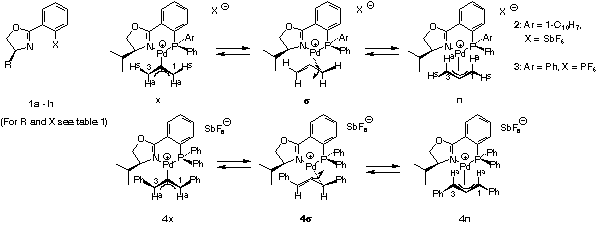
According to these experiments the Pd-C bond trans to P is clearly the kinetically more labile bond. This is further corroborated by the crystal structures (Scheme 15) of the allyl and diphenylallyl complexes. The C-Pd bond trans to P, 224 and 226 pm, is distinctly longer than the C-Pd bond trans to N, 212 and 214 pm. No doubt, the C-Pd bond trans to P is the weaker bond.
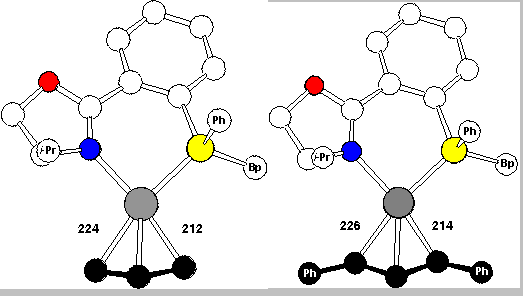
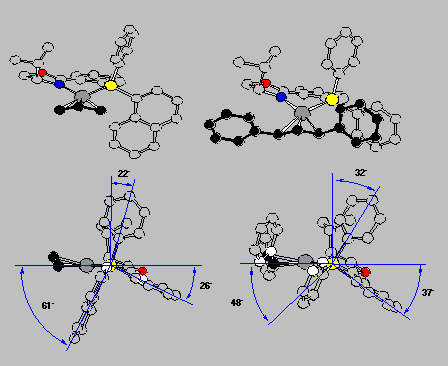
The crystal structures also explained why for the 1,3-diphenylallyl complex the exo is preferred over the endo isomer (cf. Scheme 16 and RasMol-structure). The front view, in the upper row, allows us to recognize the molecules, but the side view with the coordination plane perpendicularly arranged to the screen and P hiding N is more informative with respect to several important general points:
 Click here to view the RasMol structure
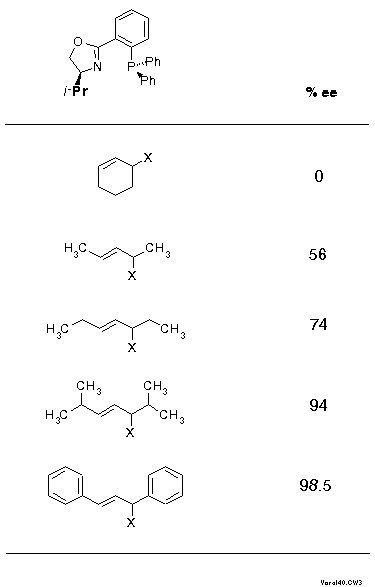
Click here to view the RasMol structureThe front view shows quite clearly that the complex fragment, the ligand, mainly provides interactions at its wings, the inner moiety is quite symmetric actually. This is schematically described in Scheme 17 as a sector rule. It appears likely that allylic systems with big substituents, such as phenyl, should display high exo-endo- and enantioselectivity, but narrow systems, with small substituents or cyclic, might give low selectivity. This is exactly what was found (cf. Scheme 18).
In Scheme 18 substrates are ordered - the isopropyl case is taken from a paper of the Pfaltz[10] - according to their "broadness" and it is quite remarkable how closely the ee values parallel the steric extension. The importance of this parameter is further underlined by NMR data of the corresponding pi-complexes: ratios of 1.8:1, 4:1, and 9:1 for the cyclohexenyl, the 1,3-dimethyl- and the 1,3-diphenylallyl-derivative (CDCl3 solution).
The assumption of an early transition state according to Bosnich is not necessarily correct. In fact, arguments in favor of a late transition state have been presented for allylic substitutions carried out with QUINAP as chiral ligand[12]. Fortunately, a late transition state, with a Pd olefins complex as model (cf. Scheme 19), is structurally quite similar to the one presented above and leads to the same conclusion: preference for substitution trans to phosphorus.