
The structures of the pterosins have led to the suggestion6 that they are derived from farnesyl pyrophosphate (1) via the same protoilludane precursor 4 which was proposed for the basidiomycete metabolite illudin-M (5).7 Evidence for this connection stems from the fact that these natural products are often isolated from the same species8,9 and that the pterosins can easily be formed by treating ptaquilosin with mild acid.10
A major obstacle to the synthesis of the pterosins is the problem of regio-selective construction of the penta-substituted aromatic ring. To date, synthetic approaches have relied heavily on classical electrophilic substitution reactions with their inherent problems of regiocontrol.11-13 In this electronic presentation, we outline the facile preparation of pterosins H, I, and Z which relies on a dipolar-cycloaddition of a cyclic carbonyl ylide dipole as the key step of the synthesis.
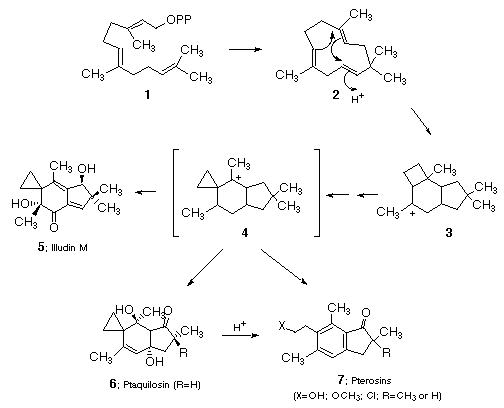
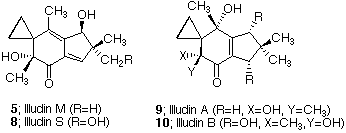
Our own interest in the pterosins evolved from our earlier work with the structurally related illudin family,14 and the strategy that evolved for our approach to the pterosin H, I, and Z was derived from that effort. Illudin M (5) and S (8) are extremely toxic sesquiterpenes produced by Omphalotus illudens, the jack-o'-lantern mushroom.15-17 Recently, two new members of this family (9 and 10) have been

isolated from a closely related fungus.18 Illudins and the related ptaquilosin (6), the aglycon of the carcinogen ptaquiloside,9 have been evaluated for antitumor activity at the NCI and show selective toxicity for human myelocytic leukemia and other carcinoma cells of various species of origin.19 As a consequence of their biological activity, it is not surprising that these compounds have received considerable attention as synthetic targets. The synthesis of illudin M and S was achieved by Matsomoto in 1971.20 Recently, Kigoshi and coworkers reported the total synthesis of (-)-ptaquilosin in 20 steps (2.9% overall yield).21
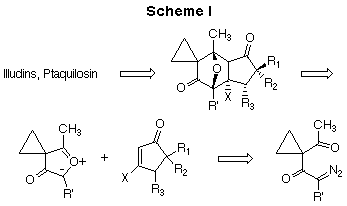
In light of the interest in this class of antitumor agents, we undertook a study designed to provide a general means for the synthesis of the core skeleton of the target molecules. In addition, because of their extreme toxicity and consequent low therapeutic index, it seemed reasonable to us to modify the basic skeleton so as to reduce cytotoxicity without compromising antitumor activity.22 Specifically, we envisioned the use of a dipolar-cycloaddition reaction of a cyclic carbonyl ylide dipole as the key step for the construction of the illudin/ptaquilosin skeleton. This strategy provides for a rapid assembly of the basic core unit of the target molecules having most of the functionality in place (Scheme I). As shown in the retrosynthetic scheme, opening of the oxy bridge of the cycloadduct would ultimately lead to the core structure of the target molecules in a highly convergent manner.
In earlier work we have described the formation of bridged oxabicyclo[3.2.1]-heptanes from the rhodium(II) catalyzed reaction of 1-diazopentanediones.23 The reaction involves the formation of a rhodium carbenoid and subsequent transannular cyclization of the electrophilic carbon onto the adjacent keto group to generate a cyclic carbonyl ylide, followed by 1,3-dipolar cycloaddition.24 Five membered ring carbonyl ylides could also be generated on treating 1-diazobutanediones with Rh(II) carboxylates.25 Thus, the Rh(II) catalyzed reaction of cyclopropyl substituted α-diazo ketones 11 and 12 resulted in cycloaddition to a variety of acyclic and cyclic alkenes. The cycloaddition proceeded readily with 1,1-dimethoxyethylene producing cycloadducts 13 and 15 in 82% and 81% yield, respectively. Reaction with α-chloroacrylonitrile gave the alternate regioisomeric cycloadducts 14 and 16 in 68% and 60% yield as a 3:1-mixture of diastereomers. The assigned regiochemistry of the products follow from their characteristic NMR spectra. When dimethyl maleate or phenylvinylsulfone were used as trapping agents, cycloadducts 17 and 18 were
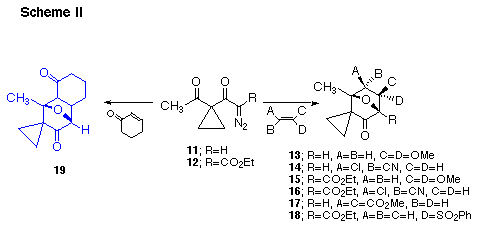
obtained as the exclusive products in 76% and 68% yield, respectively. Cyclic alkenones also participated in these tandem cyclization-cycloaddition reactions. Among the cyclic alkenes used, the reaction of cyclohexenone with 11 is noteworthy giving cycloadduct 19 (74%) as a 5:1-mixture of exo and endo isomers. The regiochemical results encountered can be rationalized on the basis of FMO considerations. For carbonyl ylides, the HOMO dipole is dominant for reactions with electron deficient dipolarophiles, while the LUMO becomes important for cycloaddition to more electron rich species.26
In our illudin and pterosin effort, the cycloaddition of 11 proceeded readily with cyclopentenone giving cycloadduct 20 as a 4:1-mixture of exo and endo isomers in 86% yield. The structure of the major diastereomer of 20 was unequivocally established by an X-ray crystal structure (see Figure 1). Isolation of exo-20 as the major stereoisomer establishes the feasibility of the planned convergent approach to the illudin family as outlined in the retrosynthetic Scheme I.
The reaction of exo-20 with 2.2 equiv of methyl iodide using potassium hexamethylsilazide as the base provided the dimethylated product 21 in 79% yield. Our expectation was that the regiospecificity of oxy-bridge cleavage could be controlled to give either the illudin or ptaquilosin skeleton based on the reaction conditions employed. Using compound 21 as a model system, we were successfully able to convert it into 22 (illudin skeleton) upon treatment with base. Further reaction of 21 with p-toluenesulfonic acid in THF produced dihydrobenzofuran 24 in 70% yield. The overall sequence of reactions can best be described as proceeding by an initial oxy-bridge ring opening followed by dehydration and a subsequent acid-catalyzed cyclopropyl ketone rearrangement.26 The facility of the process is undoubtedly related to the aromaticity gained in the final step.
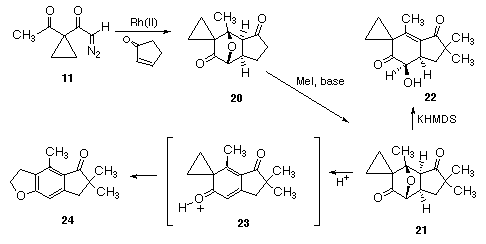
With this observation in hand, we reasoned that several members of the pterosin family would be readily accessible from the corresponding methylene derivative 25. The synthesis began by treating 21 with triphenylmethylphos-phonium bromide in the presence of sodium hydride and isolating the expected Wittig product 25 in 85% yield. By using the appropriate acid-solvent combination, we were able to obtain each of the pterosins in one step from the key reactive intermediate 27. It was even possible to isolate precursor 26 using either gentle acidic conditions or by treating 25 with n-BuLi in THF at 0oC. Thus, pterosin I (7a;
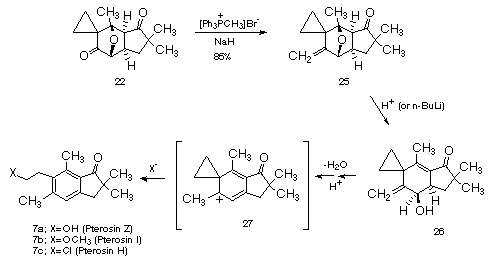
X=OCH3) was formed in quantitative yield by treating 25 with p-toluenesulfonic acid in methanol at 25[[ordmasculine]]C. Pterosin H (7c; X=Cl) was obtained in 50% yield from the reaction of 25 with HCl in dry DMF, whereas pterosin Z (7a; X=OH) was formed (50%) by treating 25 with p-toluenesulfonic acid and HCl in ethyl acetate.
In summary, a dipolar-cycloaddition strategy has been successfully applied toward the synthesis of several members of the pterosin family. Other aspects of this approach and its application to the more complex members of the illudalane class of sesquiterpenes are the subject of continuing investigations.
Acknowledgment: We gratefully acknowledge support of this work by the National Institutes of Health (CA-26751).
References and Notes
1. Hayashi, Y.; Nishizawa, M.; Harita, S.; Sakan, T. Chem. Lett. 1972, 375. Yoshishira, K.; Fukuoka, M.; Kuroyanagi, M.; Natori, S. Chem. Pharm. Bull. 1971, 19, 1491. Yoshishira, K.; Fukuoka, M.; Kuroyanagi, M.; Natori, S. Chem. Pharm. Bull. 1978, 26, 2346. Bardouille, V.; Mootoo, B. S.; Hirotsu, K.; Clardy, J. Phytochemistry 1978, 17, 275. McMorris, T. C.; Voeller, B. Phytochemistry 1971, 10, 3253.
2. Yoshishira, K.; Fukuoka, M.; Kuroyanagi, M.; Natori, S.; Umeda, M.; Morohoshi, T.; Enomoto, M.; Saito, M. Chem. Pharm. Bull. 1978, 26, 2346. Bardouille, V.; Mootoo, B. S.; Hirotsu, K.; Clardy, J. Phytochemistry 1978, 17, 275.
3. Evans, I. A. in Chemical Carcinogens, 2nd. ed.; Searle, C. E., Ed.; American Chemical Society: Washington DC, 1984; Vol. 2, pp. 1171-1204.
4. Hirono, I.; Yamada, K. in Naturally Occurring Carcinogens of Plant Origin; Hirono, I., Ed.; Kodansha-Elsevier: Tokyo-Amsterdam, 1987, pp. 87-120.
5. Ojika, M.; Wakamatsu, K.; Niwa, H.; Yamada, K. Tetrahedron 1987, 43, 5261. Van der Hoeven, J. C. M.; Lagerweij, W. J.; Posthumus, M. A.; van Veldhuizen, A.; Holterman, H. A. J. Carcinogenesis 1983, 4, 1587.
6. McMorris, T. C.; Anchel, M. J. Am. Chem. Soc. 1965, 87, 1594. McMorris, T. C.; Nair, M. S. R.; Anchel, M. J. Am. Chem. Soc. 1967, 89, 1967.
7. McMorris, T. C.; Kelner, M. J.; Wang, W.; Estes, L. A.; Montoya, M. A.; Taetle, R. J. Org. Chem. 1992, 57, 6876.
8. Yoshishira, K.; Kukuoka, M.; Kuroyanagi, M.; Natori, S. Chem. Pharm. Bull. 1978, 26, 2365.
9. Ojika, M.; Wakamatsu, K.; Niwa, H.; Yamada, K. Tetrahedron 1987, 43, 5261.
10. Niwa, H.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Hirono, I.; Matsushita, K. Tetrahedron Lett. 1983, 24, 4117. Nozoe, H.; Kobayashi, H.; Urano, S.; Furukawa, J. Tetrahedron Lett. 1977, 1381. Ayer, W. A.; McCaskill, R. H. Can. J. Chem. 1981, 59, 2150.
11. Woodward, R. B.; Hoye, T. R. J. Am. Chem. Soc. 1977, 99, 8007.
12. Ng, K. M. E.; McMorris, T. C. Can. J. Chem. 1984, 62, 1945.
13. Neeson, S. J.; Stevenson, P. J. Tetrahedron 1989, 45, 6239.
14. Padwa, A.; Sandanayaka, V. P.; Curtis, E. A. J. Am. Chem. Soc. 1994, 116, 2667.
15. Nakanishi, K.; Tada, M.; Yamada, Y.; Ohashi, M.; Komatsu, N.; Terekawa, H. Nature, 1963, 197, 292. French, A.L.; Garrettson, L.K. Clin. Toxicol., 1988, 26, 81.
16. Nakanishi, K.; Ohashi, M.; Tada, M.; Yamada, Y. Tetrahedron 1965, 21, 1231.
17. Anchel, M.; Hervey, A.; Robbins, W. J. Proc. Natl. Acad. Sci. U.S.A. 1947, 33, 171.
18. Arnone, A.; Cardillo, G.; Nasini; De Pava, O.V. J. Chem. Soc. Perkin Trans I, 1991, 733.
19. McMorris, T.C.; Kelner, M.J.; Wang, W.; Esters, L.A.; Montoya, M.A.; Taetle, R. J. Org. Chem., 1992, 57, 6876.
20. Matsumoto, T.; Shirahama, H.; Ichihara, A.; Shin, H.; Kagawa, S.; Sakan, F.; Miyano, K. Tetrahedron Lett., 1971, 2049.
21. Kigoshi, H.; Sawada, A.; Nakayama, Y.; Niwa, H.; Yamada, K. Tetrahedron Lett., 1989, 30, 1983. Kigoshi, H.; Imamura, Y.; Mizuta, K.; Niwa, H.; Yamada, K. J. Am. Chem. Soc., 1993, 115, 3056.
22. McMorris, T.C.; Kelner, M.J.; Wang, W.; Moon, S.; Taetle, R. Chem. Res. Toxicol., 1990, 3, 574. McMorris, T.C.; Kelner, M.J.; Beck, W.T.; Zamora, J. M.; Taetle, R. Cancer Res., 1987, 47, 3186.
23. Padwa, A.; Carter, S. P.; Nimmesgern, H. J. Org. Chem. 1986, 51, 1157. Padwa, A.; Carter, S. P.; Nimmesgern, H.; Stull, P. D. J. Am. Chem. Soc. 1988, 110, 2894. Padwa, A.; Fryxell, G. E.; Zhi, L. J. Org. Chem. 1988, 53, 2875; J. Am. Chem. Soc. 1990, 112, 3100.
24. For some related examples, see: Ibata, T.; Motoyama, T.; Hamaguchi, M. Bull. Chem. Soc. Jpn. 1976, 49, 2298. Maier, M. E.; Evertz, K. Tetrahedron Lett. 1988, 29, 1677. Gillon, A.; Ovadia, D.; Kapon, M.; Bien, S. Tetrahedron 1982, 38, 1477.
25. Padwa, A.; Chinn, R. L.; Hornbuckle, S. F.; Zhang, Z. J. J. Org. Chem. 1991, 56, 3271.
26. Houk, K. N.; Sims, J.; Duke, R. E.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287. Sustmann, R.; Trill, H. Angew. Chem., Int. Ed. Engl. 1972, 11, 838.
27. Schweizer, E. E.; Kopay, C. M. J. Org. Chem. Soc. 1971, 36, 1489. Danishefsky, S.; Dynak, J. Tetrahedron Lett. 1975, 79. Saalfrank, R. W.; Gundel, J.; Robmann, G.; Hanek, M.; Rost, W.; Peters, K.; von Schnering, H. G. Chem. Ber. 1990, 123, 1175.