Substrate-Directed Formation of Catalytic Metallo-Oligopeptides
Gideon Fleminger+,
Etty Kochavi* and Akiva
Bar-Nun*
+Department of Molecular Microbiology and Biotechnology
and *Department of Geophysics and Planetary Sciences, Tel Aviv University,
Tel Aviv, Israel, 69978
Abstract
Enzymatic reactions involve the association of specific amino acid residues
in the enzyme active site with certain groups
on the substrate molecule, via a series of subtle, non-covalent interactions.
We suggest that this type of specific recognition, under certain conditions,
can be utilized for substrate-induced synthesis of catalytic oligopeptides.
Incubation of a given substrate with a mixture of amino acids (and possibly
with certain metal ions) may cause their assembly into an "active-site-like"
pocket. Upon addition of a condensing agent, a (metallo)oligopeptide
should be formed, which interacts with the substrate in a specific manner.
Under certain conditions, the newly-formed molecule should act on the substrate
as an enzyme. To examine this hypothesis experimentally,
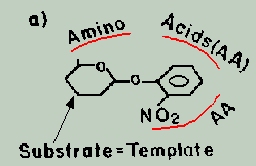
we used the substrate o-nitrophenol--D-galactopyranoside (ONPG) as a molecular
template to assemble certain amino acids and to synthesize a specific catalyst,
capable of cleaving the same substrate. This was achieved by incubation
of ONPG with a mixture of free amino acids, FeSO4 and a condensing
agent (dicyandiamide) at elevated temperatures. The reaction rate (d[product]/d2t)
increased linearly with time, indicating an acceleration regime, where
the substrate generates the formation of a catalyst. This catalyst was
purified by RP-HPLC and identified as Cys2-Fe+2
by a series of structural and chemical analyses. It was shown to catalyze
the hydrolysis of ONPG, used to assist its own formation. Based on specificity
studies, kinetic data and molecular imaging, a model for the structure
and mode of action of this catalyst is proposed.
Introduction
Association between the amino acid residues composing the active site of an enzyme and its substrate is based on a series of subtle noncovalent interactions, such as hydrogen bonds, salt bridges, hydrophobic interactions and van der Waals forces. Each of these interactions is at the range of 0.1-1.0 kcal/mol-1, the same order of magnitude of the thermal energy in solution at room temperature1. The resulting change in free energy per residue is too small to reduce the activation barrier of the reaction. However, accumulation of such changes in a cooperative manner leads to rearrangements in the enzyme-substrate complex, forming a transition state of lower energy barrier and enables the reaction to proceed under moderate conditions2. This is the reason why enzymes can bind and activate tens of thousands of substrate molecules per second and release the products when the reaction is completed.
We suggest that the unique recognition of certain amino acids of an
enzyme by the substrate may be utilized for the preparation of specific
catalytic oligopeptides. Assembly of specific amino acids around the substrate,
creating an "active site-like pocket",
followed by their condensation by a condensing agent will form an oligopeptide.
If metal ions are added, a metallo-oligopeptide
may be formed. Under certain conditions, these (metallo)oligopeptides may
serve as catalysts acting on the same substrates as illustrated in the
following scheme:
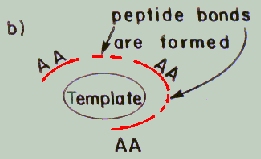
a) 1st stage: The
substrate molecule serves as a template for the formation of an active
site-like pocket, by assembling the free amino acids around it.
b) 2nd stage: Condensation
of these amino acids leads to the creation of active oligopeptides, which
might have carried out the desired chemical reactions through the same
weak forces.
To examine this hypothesis experimentally, we studied the formation
of a catalytically-active metallo-dipeptide using the substrate o-nitrophenyl--galactopyranoside
(ONPG) as a template. The hydrolysis of ONPG proceeds according to the
following scheme:
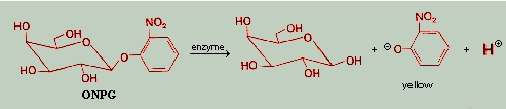
Complete details have been published elsewhere3.
Methods
The twenty naturally-occurring amino acids, at various combinations, were first incubated with ONPG to analyze their capabilities to hydrolyze ONPG without condensation4 as illustrated in Stage 1 of the above Scheme. Four of these, cysteine, tryptophan, methionine and asparagine, which have been shown to possess the highest hydrolytic activity4, were incubated with FeSO4, the substrate and dicyandiamide (DCDA), a pre-biotic condensation agent. Later, the four amino acids were replaced by cysteine alone, yielding identical results. The mixture was incubated for 2-5 days at 37-70C. At this stage, a metallo-oligopeptide was produced in a substrate-directed manner, as illustrated in Stage 2 of the above Scheme. The production of o-nitrophenol due to the hydrolysis of ONPG was monitored by the increase in the absorbance at 420 nm.
Isolation of the active species from the reaction mixture was achieved by reversed phase chromatography on a RP-18 column. Three criteria were used for identification of the reaction products:
(a) Compound(s) that have the ability to hydrolyze
the substrate.
(b) Compound(s) that are formed only in the presence
of both the substrate and the condensing agent in a time-dependent manner.
(c) Compound(s) that possess increased optical adsorbance
at 230 nm, which would indicate the formation
of peptide bonds.
Results
Isolation of the active peptides
RP-18 HPLC chromatogram, monitored at 215
nm (running buffer - 0-20% acetonitril in 0.1% trifluoroacetic acid) of
the reaction products obtained by mixtures of Cys, Met, Asn and Trp with
ONPG, FeSO4 and DCDA (red). The
blue line represents the chromatogram obtained
in absence of DCDA. Inset: Further separation of active
peak by the same column developed with 0-20% acetonitril in 0.1M phosphate
buffer, pH 7.0.
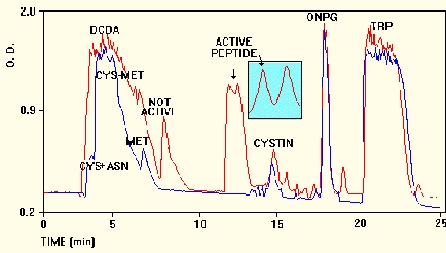
Depletion of either the substrate or DCDA
resulted in elimination of the active peak from the chromatogram. Depletion
of iron from the reaction mixture by treatment
with H2S, abolished the hydrolytic activity towards ONPG, as well as the
formation of the catalyst. Generation of the active peptide was restored
upon addition of ferrous, but not ferric, ions (10-3M). Ferrous ions by
themselves, did not possess any catalytic activity toward ONPG.
Activity of newly-formed catalyst
The oligopeptide, isolated by RP-HPLC,
catalyzed the hydrolysis of ONPG, showing an increase in the specific activity
by a factor of 104, compared to the activity of the original
mixture of amino acid. As shown below. it possessed a turn-over
number of about 1.5 h-1. At least 15 cycles for each
catalyst molecule were obtained.
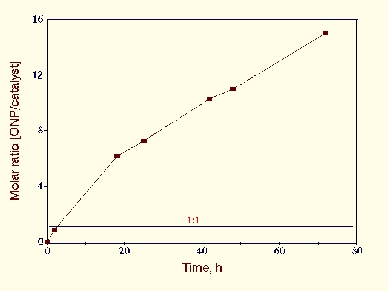
The activity was found to increase with temperature, at least up to 70C, and with Cysteine or ONPG concentrations. It was highest at the neutral pH range (pH 6.5-7.5).
Galactose (10 mM) inhibited the hydrolysis of ONPG by the active oligopeptide, but not the spontaneous hydrolysis of the substrate.
Upon replacement of ONPG by the para analog
(PNPG) or by the phosphate derivative (PNP)
the active peptide was not produced. Moreover, the active catalyst produced
by ONPG, failed to hydrolyze these analogs. From the HPLC chromatograms
other catalysts apparently were produced, which are still under investigation.
Structure of the active compound:
The purified material was subjected to a series of structural
and chemical analyses, including
amino acid analysis, mass spectrometry (FAB), flame photometry amino acid
sequencing and titration of amino- and thio- groups.
| Method | Characteristics | result |
| MS / FAB, MS / IC | molecular weight (at acidic pH) | 280 dalton |
| Amino Acid Analysis | amino acid residues (only cysteine found) | 30 nmol Cys |
| Ellman's reagent (DTNB) | thio- groups | 20 nmol |
| Florescamine and ninhydrin | amino residues | 15 nmol |
| Sequence Analysis | amino acid sequence | Cys-Cys |
| Flame photometry and atomic absorption | metal ion analysis (onliron ions found) | 30 nmol Fe |
The above data led to the following putative structure for the active compound:
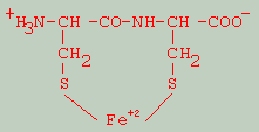
An immobilized form of Cys2Fe
To demonstrate that the formation of the active oligopeptide indeed
involves the dimerization of two cysteins in a substrate-directed manner,
we attempted to synthesize an immobilized form
of the peptide using a matrix-bound cysteine as the starting material.
Immobilized cysteine was prepared by coupling of cystine to oxirane bearing
beads (Eupergit C30N) through its amino groups5.
Reduction with -mercaptoethanol resulted the conversion of the immobilized
cystins into cysteins. These were then incubated with soluble cysteine,
DCDA, FeSO4 and ONPG for 72 h at 50C. As a result, an immobilized
form of the oligopeptide was obtained. When the immobilized Cys2Fe
was incubated with ONPG, as a substrate, high hydrolytic activity was observed.
As for the soluble form of the reaction, when the substrate ONPG was omitted
from the reaction mixture of the immobilized cysteine, no active peptide
was formed.
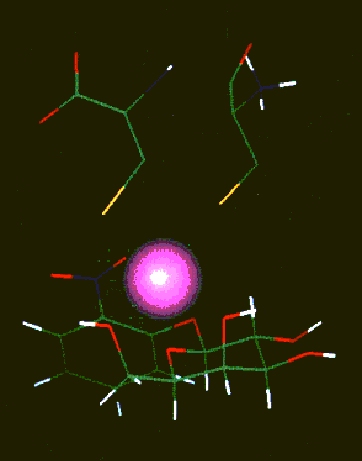
Schematic presentation of ONPG, Fe+2 and two cysteine residues
(still free). Oxygen atoms are in red and SH groups in yellow.
The model:
We propose the following model for the
formation and mode of action of Cys2Fe: A Fe+2 atom
associates with four oxygen atoms of ONPG (two of the galactose ring and
two of NO2 of the nitrophenyl ring) and the two SH groups of
the cysteins. This prevents the competing S-S oxidation of the SH groups.
The two cysteins then form a peptide bond by reaction with DCDA. Upon cleavage
of the ONPG molecule, its product leaves the Cys2Fe molecule
which is ready for association with another ONPG molecule.
Evidences for validity of the proposed model:
a. The active oligopeptide is not formed in absence
of ONPG. Instead, inactive S-S oxidized dicysteine, identified as a separate
peak by HPLC, is formed.
b. Depletion of Fe+2 ions from the reaction mixture, abolishes
the formation of the active oligopeptide.
c. Galactose inhibits the reaction in a competitive
manner.
d. Cys2Fe is not formed when ONPG is replaced
by the para analog (PNPG).
Discussion
This work demonstrates experimentally, substrate-directed formation of an oligopeptide which possesses specific catalytic activity towards the same substrate. The purified catalyst possessed specific activity which was about 104 times higher than that of a mixture of free amino acids4 and only about 2*103 times less than that of the purified -galactosidase. It's structure was determined as Cys2-Fe+2 by a series of structural and chemical analyses.
The active oligopeptide acted as a "real" enzyme, capable of successive hydrolysis of additional substrate molecules. This mechanism was found to be highly specific: the catalytic peptide formed on ONPG as a template failed to hydrolyze related substrate molecules such as PNPG and PNPP. Moreover, this peptide was not formed when ONPG was replaced in the reaction mixture by either of these two analogs. Formation of the catalytic peptide by substrate templating was found to be streospecific and anaerobically favored.
It is noteworthy that despite the huge size of some of today's enzymes, their active sites remained very small throughout their evolution. This led to the notion that "modern" enzymes evolved from small "active centers" incorporated into proteins. This idea was particularly examined with regard to metallo-enzymes which have presumably evolved from small metal-amino acids and metal-sulfide complexes6,7. Moreover, iron-sulfur complexes of the same type form an important component of several "modern" bioactive proteins (e.g. ferredoxin8 or aconitase9) and are considered to be one of the more primitive catalytic structures in nature10. The catalytic peptide Cys2-Fe+2 obtained by ONPG-directed synthesis indeed resembles very much the structure of the active sites of such iron-sulfur proteins which possess Cys2-Fe-S2 or Cys4-Fe2-S2 structures. Particularly is relevant the enzyme aconitase, where the Fe-S cluster at the active site interacts with hydroxyl groups of the substrate, citrate or isocitrate. Already in 1966, Eck and Dayhoff11 suggested that long before ferredoxin formation, the primitive cell used iron sulfide as a catalyst, probably attached to either free cysteine or an oligopeptide. Primitive enzymes of this group were found in Thermogata sp. which is considered the oldest bacteria found on Earth12. Iron sulfide, in the form of pyrite, was proposed by Wächtershäuser13.
We propose that this model should represent a
more general rule which may be utilized for the preparation of synthetic
active (metallo)oligopeptides for practically any given substrate.
References
1. Vol'kenshtein MV (1969). Induced structural correspondence of enzyme and substrate. In Enzyme physics. Plenum Press, New York-London, pp. 47-58.
2.Hammes GG (1982) Elementary steps in enzyme catalysis. In: Enzyme catalysis and regulation. Academic Press, London, pp.99-108.
3. Kochavi, E., Bar-Nun, A. and Fleminger, G. (1997). Substrate directed formation of small biocatalysts under prebiotic conditions. J. Mol. Evol. (in press).
4. Bar-Nun A, Kochavi E, Bar-Nun S (1994) An assemblage of free amino acids as a possible prebiotic enzyme. J. Mol. Evol. 39:116-122.
5. Fleminger, G., Wolf, T., Hadas, E. and Solomon, B. (1990). Eupergit C as a carrier for HPLC-based immunopurification of antigens and antibodies. J. Chromatog. 510:271-279.
6. Frieden E (1973) In: Protein-Metal interactions, eds. Friedman, M. Plenum press NY, London, pp. 1-31.
7. Riordan JF, Vallee BL (1973) The function roles of metals in metaloenzymes. In: Protein-Metal interactions. Friedman M, ed. Plenum press New York, London pp. 1-31.
8. Orne-Johnson, WH. (1973). Iron Sulphor Proteins. Ann. Rev. Biochem. 42:159-204.
9. Lauble, H. and Stout, CD. (1995). Proteins. 22:1-11.
10. Kerscher L, Oesterhelt D (1982). Pyruvate: ferredoxin oxidoreductase - new findings on an ancient enzyme. TIBS 7:371-374.
11. Eck RV, Dayhoff MO (1966) Evolution of the structure of ferredoxin based on living relics of primitive amino acid sequences. Science 152:363-365.
12. Dyer BD, Obar RA (1994) Tracing the eukariotic cells: the enigmatic smile. Colombia University press, pp.1-63.
13. Wächtershäuser G (1994) Formation of amide bonds without a condensation agent and implications for origin of life. Nature 368(6474):836-838.