
NEW ASPECTS OF CALIXPYRROLE CHEMISTRY
JONATHAN L. SESSLER,* PHILIP A. GALE, VLADIMÍR KRÁL, VINCENT LYNCH, JOHN W. GENGE, WILLIAM E. ALLEN, CHRISTOPHER T. BROWN, ANDREAS GEBAUER, NICOLAI A. TVERMOES AND PETRA I. SANSOM.
THE UNIVERSITY OF TEXAS AT AUSTIN, AUSTIN, TEXAS, 78712-1167, USA.
Introduction
Anions are ubiquitous in biological systems. They play structural roles in proteins,1 are involved in transport through membranes2 and carry genetic information (DNA and RNA are polyanions).3 There is therefore intense current interest in the complexation chemistry of these negatively charged species.4 Previous work in our research group has shown that protonated expanded porphyrins, such as sapphyrin, are excellent receptors for anions.5 The crystal structure of the fluoride complex of sapphyrin is shown in Figure 1. The anion is bound by a combination of electrostatic interactions and hydrogen bonds resulting in a very stable complex. Although these receptors molecules are effective anion binding agents, they are challenging to prepare. Thus began the search for other easier-to-make polypyrrolic macrocycles that might serve as alternative receptors for anionic species.
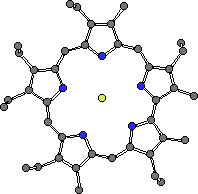
Figure 1. The fluoride complex of a diprotonated sapphyrin. The F- is 'chelated' within the sapphyrin core. There is a second counter anion (PF6-) that is not proximate to the macrocycle skeleton.
CHIME CRYSTAL STRUCTURE: sapphyrin fluoride complex
Our search for a new anion binding platform ultimately led us to the octaalkylporphyrinogens. Simple porphyrinogens, bearing four or fewer meso-substituents are naturally occurring colourless macrocycles that contain four pyrrole rings linked in the 2, and 5 positions via sp3 hybridised carbon atoms. Such systems have attracted a lot of attention, primarily because they may be oxidised to form porphyrins. However meso-octaalkylporphyrinogens have attracted much less attention, as they are stable materials that are resistant to such oxidations (Scheme 1).
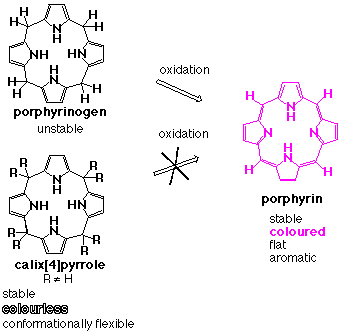
Scheme 1.
We felt that these materials were misnamed and might better be referred to as calix[4]pyrroles, drawing a parallel with their phenolic cousins, the calixarenes, and emphasising the interesting conformational properties of macrocycles. To the extent this renaming had merit, we posited that such systems could function well as anion binding agents (vide infra).
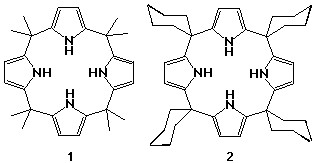
Calixpyrroles were first synthesised in 1886 by Baeyer.6 He condensed pyrrole and acetone in the presence of hydrochloric acid and obtained a white crystalline material which later proved to be meso-octamethylcalix[4]pyrrole 1. The reaction was refined over the following century to a stage where 90% yields of the macrocycle are readily obtainable without the need for any form of templation or for high dilution conditions. Other calix[4]pyrroles have been synthesised since then including meso-tetraspirocyclohexylcalix[4]pyrrole 2, formed from the condensation of cyclohexanone and pyrrole.7
The transition metal coordination chemistry of deprotonated meso-octaalkylcalix[4]pyrroles has been studied by Floriani and co-workers. Many metallic complexes have been prepared from meso-octaethylcalix[4]pyrrole, which is first deprotonated, and then complexed to the metal ion.8 These complexes have interesting electrochemistry, some forming so-called "artificial porphyrins" when oxidized.9
By contrast, prior to our interest in the subject, the anion coordination chemistry of these macrocycles had not been studied.
Results and Discussion
Surprisingly, the crystal structure of 1 had not been eludicated in the over one hundred years since it was first synthesised. Crystals of 1 and 2 were grown by slow evaporation of acetone and dichloromethane solutions of the ligands respectively. Single crystal X-ray diffraction analysis revealed that both compounds adopt the 1,3-alternate conformation in the solid state (Fig. 2).10
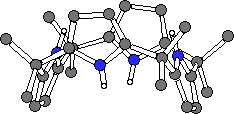 1
1 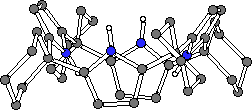 2.CH2Cl2
2.CH2Cl2
Figure 2. The X-ray crystal structures of 1 and 2.
CHIME CRYSTAL STRUCTURES: 1 and 2.CH2Cl2.
Stability constant determinations were made using 1H NMR titration techniques and subsequent analysis using the EQNMR computer program.11 The findings, summarised in Table 1, reveal that both compounds 1 and 2 are not only effective 1:1 anion binding agents in solution, but are also selective, showing a marked preference for F- relative to other putative anionic guests (viz. Cl-, Br-, I-, H2PO4- and HSO4-).
Link to Table 1: stability constants of anion complexes of compounds 1 and 2.
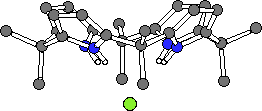
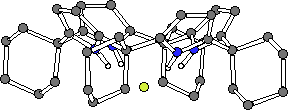
Crystals of the tetrabutylammonium chloride complex of calixpyrrole 1 and the fluoride complex of 2 were obtained by slow evaporation of a dichloromethane solution of these ligands containing an excess of the halide salt. The crystal structures revealed that in both cases the calix[4]pyrrole ligand adopts a cone-like conformation such that the four NH protons can hydrogen bond to the halide anion. While these two structures are similar, in the case of the chloride complex the nitrogen-to-anion distances are in the range 3.264(7) - 3.331(7) Å, while inthe corresponding fluoride complex they are 2.790(2)Å (Fig. 3b) (the four pyrrole groups are equivalent by symmetry). As a result, in these two complexes the chloride and fluoride anions reside 2.319(3) Å and 1.499(3) Å above the N4 root mean square planes of calixpyrroles 1 and 2, respectively.
 1.Cl-
1.Cl-
 2.F-
2.F-
Figure 3. The X-ray crystal structures of 1.Cl- and 2.F- (tetrabutylammonium cations and solvent omitted for clarity).
CHIME CRYSTAL STRUCTURES: chloride complex of 1 and the fluoride complex of 2.
Variable temperature 1H NMR spectroscopic studies were carried out on dichloromethane-d2 solutions of 1 measured in the absence and presence of fluoride anions. In the presence of three equivalents of tetrabutylammonium fluoride the meso-methyl resonance splits into two as the temperature is lowered (Fig. 4), whereas in the absence of fluoride anions, there is no significant change in the 1H NMR of compound 1. This splitting may be due to the calixpyrrole adopting a cone conformation in solution when bound to fluoride. The meso-methyl groups will then be arranged in either axial or equatorial positions and therefore resonate at different frequencies. At room temperature this is not observed because the complexation-decomplexation rate is fast compared to the NMR timescale. However as the temperature is lowered this process is slowed and the cone conformation becomes detectable.
Surprisingly, the pyrrole NH resonance also split into two as the temperature was lowered in the presence of fluoride (Fig. 5). One possible cause for this would be coupling to the 19F nucleus of the bound fluoride anion to the NH protons. To test this supposition, a 19F NMR spectrum of the fluoride complex was acquired at 193 K. The bound fluoride ion resonated as a quintet (with a coupling constant of 39.5 Hz) confirming the coordination coupling effect.
The binding abilities of the calix[4]pyrroles are not limited to anions and cations. Coordination of neutral species has also been achieved using meso-octamethylcalix[4]pyrrole 1. 1H NMR titration experiments in benzene-d6 and subsequent analysis of the titration curves using EQNMR11 revealed that meso-octamethylcalix[4]pyrrole forms complexes with neutral species including short-chain alcohols, amides and other oxygen containing neutral species, with moderate stability constants in benzene-d6. The best data fit was achieved assuming 1:1 stoichiometries in solution. A trend is evident across the alcohol and amide series, in which the stability constant decreases with increasing steric bulk around the oxygen atom (Table 2), although other factors also play a role.
Link to Table 2: stability constants of neutral guest complexes of compound 1.
Single crystals of meso-octamethylcalix[4]pyrrole 1 coordinated to methanol were prepared by slow evaporation of dichloromethane solution of 1 in the presence of the alcohol. In contrast to the solid state structures of the anion complexes of 1 and 2, the methanol complex of 1 adopts a 1,3-alternate conformation (Fig. 6). Two methanol molecules are coordinated to the calixpyrrole via hydrogen bonds from two pyrrole moieties to each methanol with Npyrrole-OMeOH = 3.155(4) Å. Single crystals of the DMF complex of 1 have also been prepared. Again, two DMF molecules are coordinated to a single calix[4]pyrrole via two hydrogen bonds to each molecule, however in this case the calix[4]pyrrole adopts a 1,2-alternate conformation wherein each DMF molecule is coordinated to adjacent pyrrole moieties (Npyrrole-ODMF = 2.908(2) and 2.924(2) Å) (Fig. 6). The DMF molecules lie over the plane of a pyrrole ring (to which they are not hydrogen bonded) at a distance of 3.363(3) Å, suggesting that a pi-pi interaction is stabilising the 1,2-alternate conformation of the calix[4]pyrrole.
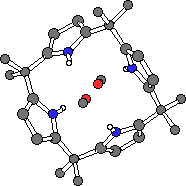 1.2MeOH
1.2MeOH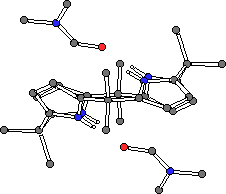 1.2DMF
1.2DMF
Figure 6. Single crystal X-ray structures of the methanol (left) and DMF (right) complexes of meso-octamethylcalix[4]pyrrole 1.
CHIME CRYSTAL STRUCTURES: methanol complex of 1 and the DMF complex of 1.
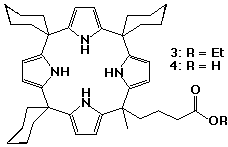
The introduction of a carboxylate group to a calix[4]pyrrole has produced an anionic calixpyrrole with interesting self-assembly properties.13 A "meso-hook" calix[4]pyrrole monoester 3 was synthesised by co-condensing 4-methylacetylbutyrate, cyclohexanone and pyrrole. After column chromatography, the mono-ester was isolated from a mixture of calixpyrroles containing different numbers of ester functional groups, in 12% yield. Subsequent hydrolysis of 3 yielded the monoacid 4.
X-ray quality crystals of the calixpyrrole carboxylate 4 were obtained by slow evaporation of a dichloromethane solution of 4 in the presence of excess tetrabutylammonium fluoride hydrate. Interestingly, the crystals did not contain any fluoride anions, but instead consisted of the tetrabutylammonium calix[4]pyrrole carboxylate salt. The structure of the salt revealed that the calixpyrrole carboxylate self-assembles in the solid state with the carboxylate of one calixpyrrole bound to the pyrrolic array of an adjacent calixpyrrole and vice-versa, forming a dimeric cyclic structure. The calix[4]pyrrole adopts the cone conformation with four hydrogen bonds from the pyrrole groups to the bound carboxylate oxygen.
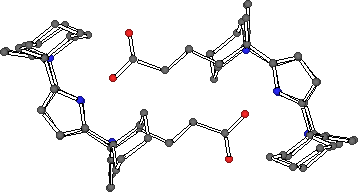
Figure 7. X-ray crystal structure of the self-assembled calixpyrrole dimer 4.4 (tetrabutylammonium cations omitted for clarity).
CHIME CRYSTAL STRUCTURE: self-assembled calixpyrrole dimer
A ROESY NMR spectrum of the trimethylammonium salt of 4 recorded in dichloromethane-d2 provided evidence for aggregation in solution (Fig. 8). Two resonances were observed between 7.0 and 7.5 ppm, corresponding to non-complexed pyrrole NH protons. Another resonance was observed at approximately 11 ppm, corresponding to bound pyrrole NH protons, in exchange with the unbound NH resonances, indicating aggregate formation in solution. In this salt, the counter cation may also form hydrogen bonds to the carboxylate moieties thereby favouring the monomeric species in solution. The tetrabutylammonium salt of 5 displayed pyrrole NH protons at around 11 ppm (only) in dichloromethane-d2. Upon addition of methanol to the NMR solution, the resonance at 11 ppm vanishes presumably reflecting the break up of the aggregates caused by the presence of a more competitive hydrogen bonding solvent. The dimer could also be observed using FAB MS techniques. To the best of our knowledge, the self-assembling calix[4]pyrrole system represents the first and only example of the self-assembly of purely anionic sub-units.
Link to Figure 8: ROESY NMR spectrum of the trimethylammonium salt of 4.
Several novel calix[4]pyrrole molecules containing functional groups appended to the carbon- or C-rim of the calix[4]pyrrole have been synthesised.12 Two strategies were pursued in the synthesis of these materials. Firstly, beta-octamethoxy-meso-tetraspirocyclohexylcalix[4]pyrrole 5 was prepared by condensating 3,4-dimethoxypyrrole15 with cyclohexanone in glacial acetic acid. The resulting calixpyrrole was isolated after column chromatography in 8% yield. The second strategy was to modify the C-rim of a pre-synthesised calix[4]pyrrole. Meso-octamethylcalix[4]pyrrole was dissolved in dry THF and cooled to -78oC. A solution of n-butyllithium in hexanes (4.0 equiv.) was added dropwise to the calixpyrrole solution followed by 4.0 equivalents of ethyl bromoacetate. Purification by column chromatography afforded two isolatable products, a C-rim monoester 6 (formed in 26% yield) and a diester 7 (3% yield). Surprisingly 1H and 13C NMR experiments showed that the diester 7 present in the fraction collected was a single isomer. The 1,3-substitution pattern was elucidated by single crystal X-ray diffraction analysis although the structure obtained would not refine adequately. Beta-octabromo-meso-octamethylcalix[4]pyrrole 8 was synthesized in 90% yield by reaction of meso-octamethylcalix[4]pyrrole with N-bromosuccinimide in dry refluxing THF. Single crystals of 8 were obtained by slow evaporation of a dichloromethane solution of the ligand. The structure reveals that compound 8 exists in a chair-like flattened 1,2-alternate conformation in the solid state (i.e., the dihedral angles between pyrrole rings and plane through the calixpyrrole meso-carbon atoms are 66.8o, 5.8o, -66.8o and -5.8o).
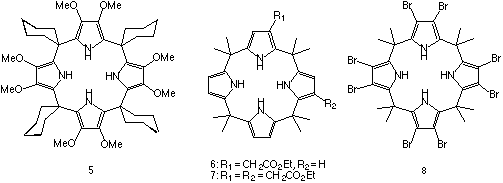
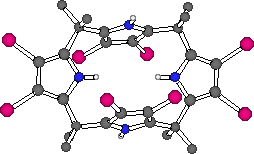
Figure 9. The X-ray crystal structure of compound 8.
CHIME CRYSTAL STRUCTURE: compound 8.
The solution anion binding properties of the beta-octamethoxy derivative 5 and the beta-octabromo derivative 8 have been studied using 1H NMR titration techniques in dichloromethane-d2 solution (Table 3). Compound 5 has lower stability constants than the corresponding "beta-free" analogue 2, presumably due to the electron donating effects of the methoxy groups that serve to decrease the acidity of the pyrrole NH protons thereby lowering the stabilty of the calixpyrrole-anion complex formed. Conversely, compound 8 has higher stability constants with anions than its "beta-free" analogue (compound 1) due to the electron withdrawing effect of the bromine substituents and the consequent increase in the acidity of the calixpyrrole.
Link to Table 3: stability constants of compounds 5 and 8 with anionic guest species.
It occured to us that a p-tert-butylcalix[n]arene could be used as a template around which a calix[n]pyrrole could form.16 Such a strategy may eventually lead to the synthesis of expanded calix[n]pyrroles where n > 4. As a first step towards this goal, p-tert-butylcalix[4]arene tetramethyl ketone was condensed with pyrrole in the presence of methanesulfonic acid to afford the cylindrical calix[4]arene-calix[4]pyrrole pseudo dimer 9 in 32% yield.
Single crystals of 9 were grown by slow evaporation of a dichloromethane solution of the ligand. The structure, as expected, shows the calixarene adopting a cone conformation. The two pyrrole NH groups form hydrogen bonds to the phenolic oxygen atoms at the lower rim of the calixarene.
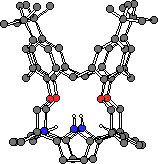
Figure 10. The X-ray crystal structure of compound 9.
CHIME CRYSTAL STRUCTURE: compound 9.
The hydrogen bonding between the pyrrole NH groups and phenolic oxygen atoms is maintained in solution as evidenced by the 1H NMR spectrum. The pyrrole NH protons resonate at 11.22 ppm indicating that they are deshielded due to their participation in the hydrogen bond. Addition of polar solvents such as methanol-d4 or anions such as fluoride does not disrupt the hydrogen bonding array indicating the molecule is adopting a cylindrical conformation in solution.
Link to Figure 11: 1H NMR of compound 9.
Conclusions
In summary, we have shown that calix[4]pyrroles are effective and selective anion and neutral binding agents in both the solid and solution states, the nature of the bound guest influencing the conformation of the receptor. The anion binding ability of these receptors can be tuned by appending different groups to the carbon or C-rim of the calixpyrrole. Carboxylate-appended calixpyrroles self-assemble forming dimeric structures, the first and only example of purely anionic subunits that undergo self-assembly. We are continuing to synthesise novel calixpyrrole species and study their versatile coordination properties.
Acknowledgements
Support from the NIH and NSF (grants AI33577 and CHE 9122161, respectively to J.L.S.) is gratefully acknowledged. P.A.G. wishes to thank the Fulbright Commission for a post-doctoral research scholarship. W.E.A. is an American Cancer Society post-doctoral fellow.
References
1 P. Chakrabarti, J. Mol. Biol. 1993, 234, 463-482.
2 M. A. van Kuijck, R. A. M. H. van Aubel, A. E. Busch, F. Lang, F.G.M. Russel, R. J. M. Bindels, C. H. van Os and P. M. T. Deen, Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5401.
3 B. J. Calnan, B. Tidor, S. Biancalana, D. Hudson and A. D. Frankel, Science 1991, 252, 1167.
4 a) F. P. Schmidtchen, Nachr. Chem., Tech. Lab. 1988, 36, 8; b) H. E. Katz, in Inclusion Compounds; ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Academic Press, New York, 1991, vol. 4, p 391; c) B. Dietrich, Pure Appl. Chem. 1993, 65, 1457; d) P. D. Beer, Chem. Commun. 1996, 689 and references cited therein.
5 J. L. Sessler and A. K. Burrell, Top. Curr. Chem. 1991, 161, 177.
6 A. Baeyer, Ber. Dtsch. Chem. Ges. 1886, 19, 2184.
7 W. H. Brown, B. J. Hutchinson and M. H. MacKinnon, Can. J. Chem. 1971, 49, 4017.
8 D. Jacoby, C. Floriani, A. Chiesi-Villa, C. Rizzoli, J. Chem. Soc. Chem. Commun. 1991, 790.
9 C. Floriani, Chem. Commun. 1996, 1257.
10 P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc. 1996, 118, 5140.
11 M. J. Hynes, J. Chem. Soc., Dalton Trans. 1993, 311.
12 W. E. Allen, P. A. Gale, C. T. Brown, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc. 1996, 118, 12470.
13 J. L. Sessler, A. Andrievsky, P. A. Gale and V. Lynch, Angew. Chem. Int. Ed. Engl. 1996, 35, 2782.
14 P. A. Gale, J. L. Sessler, W. E. Allen, N. A. Tvermoes and V. Lynch,Chem. Commun. 1997, 665.
15 A. Merz, R. Schropp and J. Lex, Angew. Chem. Int. Ed. Engl. 1993, 32, 291.
16 P. A. Gale, J. L. Sessler, V. Lynch and P. I. Sansom, Tetrahedron Lett. 1996, 37, 7881.
E-mail regarding this article may be sent to: JLS, or PAG.
Produced on an Apple Macintosh Powerbook 1400cs. This page should be viewed with Netscape 3.0.1.
![]()