Centre for Molecular Architecture, Central Queensland University, Rockhampton, Queensland, 4702, Australia.
Abstract: 8,8-Dimethylisobenzofulvene is generated in
situ and trapped with the heterodienophiles thiophosgene,
diethyl oxomalonate and tosyl cyanide to produce [4p+2p]
cycloadducts; ring-opening and rearrangement reactions of the
keto and thione adducts yielded novel indeno[1,2-c]furans and
indeno[1,2-c]thiophens while the nitrile adduct could be converted
to the related 2azanorbornan-3-one without change to the
bicyclic ring-system. None of these products served as a precursor
to 8,8dimethylisobenzofulvene. Graphical Abstract
1. Introduction
Cram and his group at UCLA have recently described a new technique for the stabilisation of reactive species by encapsulation within calixarene-based hosts [2]. We have conducted much research on the chemistry of isobenzofulvenes and used trapped adducts to evaluate periselective outcomes with various cyclic polyenes [3]. The blue-coloured, monomeric isobenzofulvene has been prepared by a flash vacuum pyrolysis technique and found to be stable up to -100 oC [4] The Cram technology appeared to offer a unique opportunity to stabilise isobenzofulvene systems at room temperature and allow their study by NMR techniques. It soon became apparent that existing thermal or photochemical routes to isobenzofulvenes involved large sized precursors or high temperature conditions unsuitable for encapsulation studies. Accordingly, we sought new small-sized precursors for the photochemical generation of isobenzofulvenes. We have recently conducted a study on the host, guest size compatibility using computation methods, and can now predict the appropriate guest dimensions for successful hemicarcerand formation [5].
We report herein our synthetic studies on the preparation of
heteroalicyclic precursors which have the potential to expel small
stable molecules and yield isobenzofulvenes.
1.1. Earlier Approaches to the Synthesis of Isobenzofulvenes
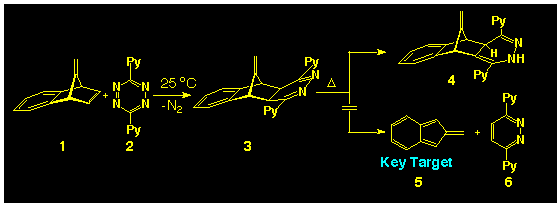
Although known to theoreticians for over three decades
[6], isobenzofulvene 5
has resisted the wiles of the organic chemist and still remains
to be identified as a monomeric species. To date, the only syntheses
of the parent isobenzofulvene 5 have elimination processes
[4,7], and while it has been possible
to produce [4p+2p]
cycloadducts when generated in the presence of dienophiles such
as N-methyl maleimide, only dimeric products are formed
in the absence of trapping agents. Although successful in producing
8-monosubstituted-isobenzofulvenes and 8,8-disubstituted isobenzofulvenes
[8], application of the s-tetrazine-induced
fragmentation of 7-methylenebenzo-norbornadiene 1 failed
to generate the parent isobenzofulvene and only the 1,4-dihydropyridazine
4, formed by rearrangement of the intermediate 1,2-dihydropyridazine
3 (Scheme 1). It is now known
that such dihydropyridazine rearrangements can be suppressed in
the presence of triethylamine [9] but
application of this new finding has not been attempted at this
point in time.
Following the successful isolation of oxylylene
8 via the matrix photolysis of the diazo compound
7 [10] (Scheme
2), an attempt was made to synthesize the diazo compound
9b, as a photochemical precursor to the 8,8-dimethyl substituted
isobenzofulvene 10, with the intention of extending this
idea to the generation of the parent compound 5 from 8a.
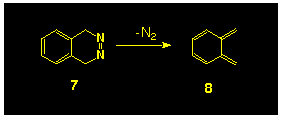
This idea, however, was not realised as attempted hydrolysis
of the intermediate hydrazide 12 resulted in ring cleavage
to yield the substituted indene 13 (Scheme
3) [11]. Clearly this type
of rearrangement could not occur in the parent system; further,
more recent studies have shown that carbamate esters of this type
can be hydrolysed at low temperature using BBr3,
however neither of these features has been explored.
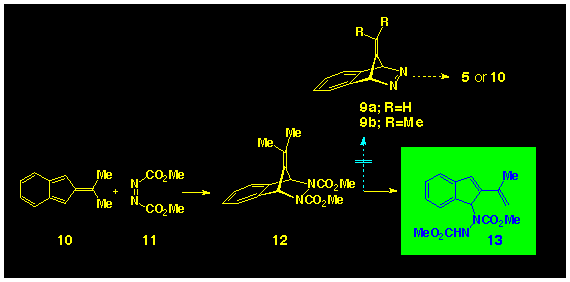
In view of the recent success in generating isoindene 15 via
the photochemical bisdecarbonylation of a-diketone
14 [12] and of spirocyclopropylisoindene
17 from irradiation of adiketone
16 [13] the diketone 19
presented itself as a promising precursor to isobenzofulvene
(Scheme 4). However, irradiation of
19 lead to a photoinduced isomerisation and production
of the enolone 22 as the sole photoproduct.
Thus, we turned our attention to the heterocyclic compounds 23-25
(Scheme 5) as possible precursors for
isobenzofulvene, and the present study describes some of our synthetic
endeavours in their production.
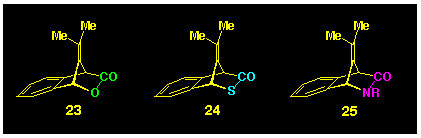
1.1 Background to the selection of the target molecules 23-25
The use of blactones as photochemical
precursors to thermally labile systems has been established.
The cheletropic loss of carbon dioxide from the b-lactone
26 is now a well developed route to cyclobutadiene 27
[14] (Scheme
6, boxed section), and was used as the precursor in Cram's
study which tamed cyclobutadiene [2]. Thus, the selection lactone
23 was justified on this basis. By analogy, it was of
considered of interest to investigate the photochemical activity
of the related thiolactone 24.
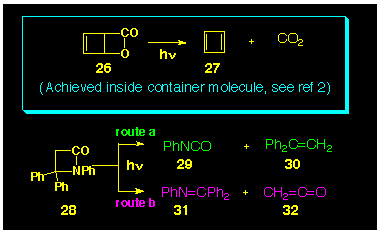
Although the cheletropic loss of isocyanic acid (HNCO) from blactams is not presently known, the photochemical loss of phenyl isocyanate (PhNCO) from the substituted b-lactam 28 has been reported [15] (Scheme 6, route a). In this system, however, photochemical cleavage of the carbonyl-to-nitrogen bond has also been observed [15] resulting in the production of imine 31 together with ketene 32 (Scheme 6, route b).
Because of the synthetic accessibility of 8,8-dimethylisobenzofulvene
10 it was decided to use these as model systems to check
the feasibility of these heterocyclic compounds as photochemical
precursors to isobenzofulvenes 10. It is well established
that 8,8dimethylisobenzofulvene 10 can be readily
produced from 33 by the stetrazine route (Scheme
1), which, in the presence of the heterocyclic dienophile
was expected to yield the adducts 35, 37, 39
which were to be modified to give the desired carbonyl compound
23-25 respectively (Scheme 1).
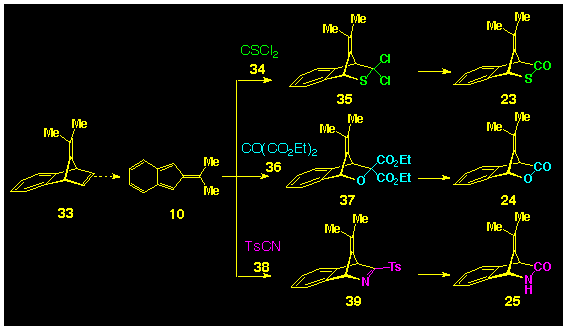
Thiophosgene 34 is an established dienophile and has been
reported to form [4p+2p]
products with cyclopentadiene 14 [16],
spirocyclopropyl cyclopentadiene 15 [17]
and 9-methyl anthracene 16 [18].
Such adducts are known to be readily hydrolysed to the corresponding
bicyclic thiolactone [19]. In addition,
several ketone have been observed to participate in Diels-Alder
cycloadditions and their reactivity is increased when electron-withdrawing
groups are attached to the carbonyl group, thereby favouring the
proposal for the use of diethyl oxomalonate 36 in this
role [19]. Ruden and Bonjouklian[20] have proposed the use of diethyl oxomalonate as a
synthetic equivalent for carbon dioxide and have reported that
the gem-diester groups can be transformed to a ketone using
the Curtius reaction. Nitriles are less common as dienophiles
and most work to date has utilised sulfonyl cyanides such as 38
owing to their high dienophilicity [21].
Indeed, the role of simple nitriles as cycloaddition reagents
has only been reported this year [22].
Facile hydrolysis of tosylcyanide adducts to lactams has
also been described [19].