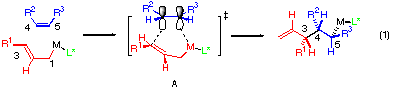
| [Molecules: 16] [Related articles/posters: 023 114 056 065 002 ] |
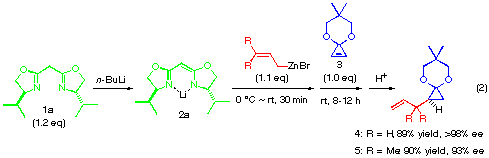
The extension of these findings subsequently led us to discover that an allylic zinc reagent bearing an anionic bis-oxazoline ligand takes place with excellent enantioselectivity (eqn. 2).[2] For instance, the reaction of the cyclopropenone acetal 3 with an allylic zinc bromide in the presence of one equivalent of the anionic bis-oxazoline ligand 2a proceeded smoothly at room temperature to afford the cyclopropanone acetal 4 in high yield with the enantioselectivity better than 98% ee. On the other hand, in the reaction of substituted allylic zinc reagents, e.g., cinnamyl zinc reagent, both the enantioselectivity and the 1,2-diastereoselectivity for the newly formed C-C bond eroded rather mysteriously. Interpretation of the mixed success and the resolution of these problems by analysis with pencil and paper appeared to be difficult since very little was known of the nature of the olefin carbometalation reactions. We thus felt it necessary to obtain molecular-level understanding of the stereochemistry of allylmetalation of olefins by computational analysis. By following the protocol we established previously for carbometalation reactions with organolithium and copper reagents,[3] we first analysed the reaction with ab initio calculations for simple models, and then with semiempirical methods for larger systems. By taking advantage of the capability of electronic publication to handle 3D pictures, we present herein our computational studies graphically displaying various transition structures in olefin allylmetalation reactions.
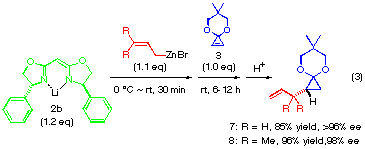
The addition of a substituted allylic zinc reagent creates an issue of mutual face selection for the two olefinic faces of both reactants, that is, 1,2-diastereoselectivity concerning the newly formed C-C bond. The reaction of substituted allylic zinc reagents 9, 10 and 11 were thus examined in the presence of anionic bis-oxazoline ligand (2a or 2c) as shown in eqn. (4). As summarized in Table 1, the diastereoselectivity for crotylzinc reagent 9, cinnamyl reagent 10 and perhydrocinnamyl reagent 11 was moderate (72:28, 73:27 and 83:17, respectively) and when the anionic bis-oxazoline ligands 2a and 2c was used. The use of tert-butyl substituted ligand as in 11 did not improved the 1,2-diastereoselectivity (entries 3 and 4) while it greatly improved the enantioselectivity from 62 to 97%.
The 1,2-diastereoselectivity of the addition of a metal enolate or an allylic metal reagent to a carbonyl group conventionally calls for chair and boat transition states.[4] On the other hand, we have reported previously in a preliminary form that the allylmetalation of isolated C-C double bond and triple bond may proceed through a single half chair transition state. We thus suspected that the flexibility of the half chair transition state may be the reason for the erosion of the selectivity, and started to investigate the details of the transition state first at the ab initio level for simplified model systems and then at the semiempirical level for more realistic models.
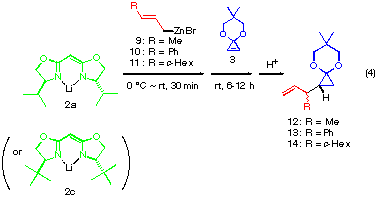
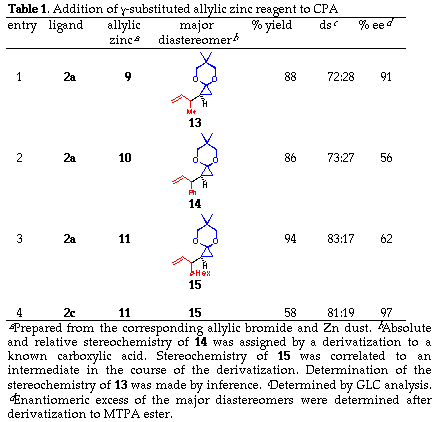
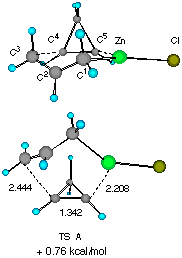
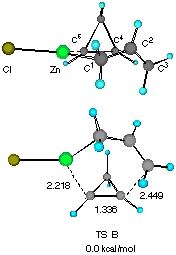
Figure 1 Two configurationally different transition structures of the addition of allylzinc chloride to cyclopropene at the HF/3-21G level
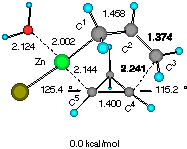
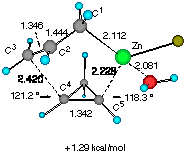
Solvent effects were then investigated by adding one molecule of water (a model for an ethereal solvent) on the metal so that coordinative saturation is achieved (Figure 2). It is notable that solvation does not affect much the gross molecular geometry of the TSs, only elongating the forming C4-C5 and C1-Zn bond by 1-2%. Apparently, the electronic background that determines the basic half chair conformation (vide supra) is strong enough not to allow large structural perturbation by a solvent molecule. The relative energy of the two TSs was however raised slightly to 1.29 kcal mol-1, suggesting that solvation may significantly affect the stereoselectivity.[10].
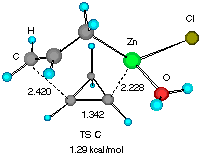
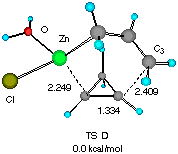
Figure 2 Two configurationally different transition structures of the addition of allylzinc chloride to cyclopropene in the presence of a H2O molecule at the HF/3-21G level
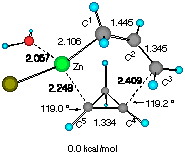
Finally, the reaction of water-solvated crotylzinc chloride with cyclopropene was studied. We also found two TSs of nearly equal energy as shown in Figure 3. The calculated energy difference (0.369 kcal mol-1) at this level of approximation of solution reactions should be viewed as negligible, yet some important characteristics were noted. First, only the half-chair TSs are available for the reaction as in the previous models. Secondly, there may occur some torsional strain for the forming C3- C4 bond in TS E (indicated by an arrow) which may slightly destabilize this transition structure. In TS F, on the other hand, the short distance (2.593 angstrom) between the two asterisked hydrogens suggests that TS F would be significantly destabilized if H** is replaced by an alkoxy group as in our substrates CPA 3.
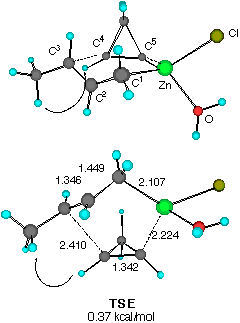
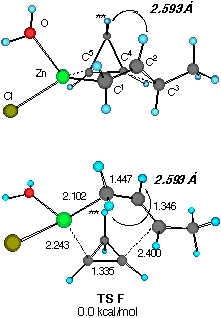
Figure 3 Two configurationally different transition structures of the addition of trans-crotylzinc chloride to cyclopropene in the presence of a H2O molecule at the HF/3-21G level
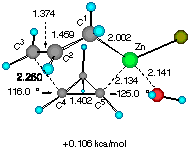
 ��������������������
��������������������
 ��������������������
��������������������Figure 4 Comparison of HF and MNDO transition structures of the addition of allylzinc chloride to cyclopropene in the presence of a H2O molecule
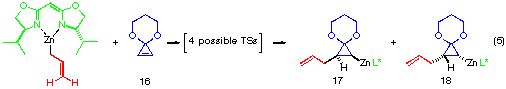
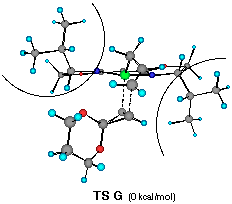
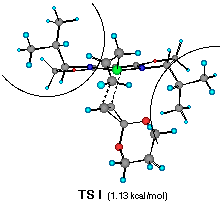
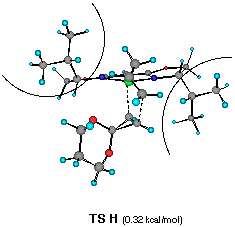
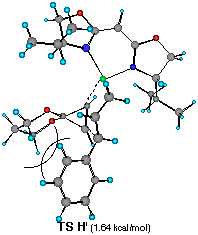
The model reaction studied by the MNDO method is the allylzincation of cyclopropenone acetal 16, lacking the gem-dimethyl groups in 3, which gives rise to two enantiomers 17 (experimental major enantiomer) and 18 (minor isomer ) (eqn. 5). Two conformationally isomeric TSs are available for each enantiomer, and in Figures 5a and b are shown these four possible TSs. The TSs are fully optimized by the MNDO method without any structural restriction. The values in parentheses refer to the computed relative heat of formation (Figure 5a). TS G and TS H differ from each other for the orientation of the allylic moiety relative to the ligand and produces the enantiomer 17. Similarly, TS J and TS I lead to the minor isomer 18. TS G and TS H are found to be ca. 1 kcal mol-1 preferred than TS I and TS J. The calculated enantioselectivity from the energy difference is 91:9 at 20 oC and this result reproduces the experimental trend (>99:1). Topological relationship between the ligand isopropyl groups and the acetal group is illustrated in Scheme 2, showing that the enantioselectivity with respect to the cyclopropenone acetal olefin is determined by steric interactions between the bis-oxazoline substituents and the bulky acetal group. An alternative way of viewing the matter, one may perceive that the six-centred TS of the allylzincation is recognized by the chiral 'cleft' formed by the two isopropyl groups.
One notable structural feature in these TSs is the tetrahedral coordination geometry for the metal, which is well protected by the bulky ligand. The lack of chelation between the metal and the acetal oxygen is noteworthy. These structural features agree well with the experimental observations that the solvent basicity (e.g., THF vs. CH2Cl2) or the presence of very basic ligand (e.g., HMPA) did not affect the selectivity much . This latter observation stand in contrast to the fact that stereochemistry of the simpler allylic zinc reagents (e.g., allylzinc bromide) is highly susceptible to solvent effects. [10]
 ��������������������
��������������������
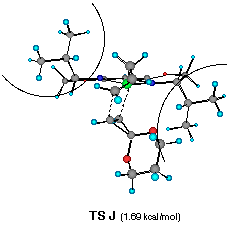 ��������������������
��������������������Figure 5a Front views of four possible transition structures of the addition of the chiral allylzinc reagent to cyclopropenone acetal and relative heat of formation. The dotted lines depict the forming C-C and C- Zn bonds. The curved lines represent the steric bulkiness of the isopropyl substituents of the chiral bis-oxazoline ligand
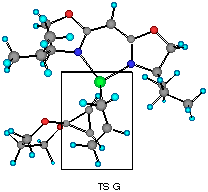
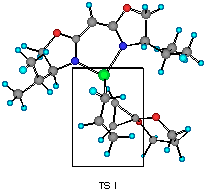
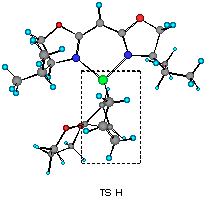
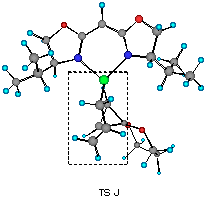
Figure 5b Top views of four possible transition structures of the addition of the chiral allylzinc reagent to cyclopropenone acetal. In the solid boxes are contained six-membered TS of one of the two configurations. The dotted boxes contains six-membered TS of another configuration
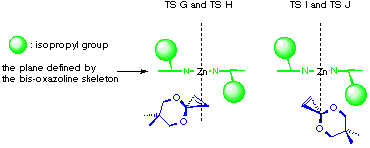
Scheme 2 Schematic of the front views of TS G, H, I and J.
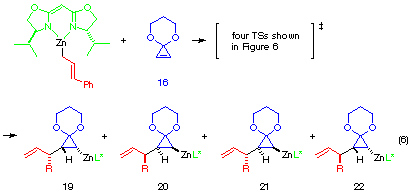
With a substituted allylic zinc reagent such as cinnamylzinc reagent, the degeneracy of the product in Figure 5 disappears and each four TSs lead to four different products, two enantiomeric and two diastereomeric pairs. In Figure 6 are shown the four possible transition structures of the reaction of CPA 16 with a cinnamylzinc possessing chiral ligand 2a (eqn. 6).
 ��������������������
��������������������
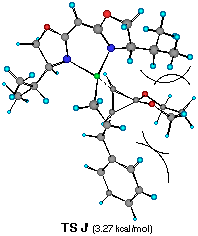 ��������������������
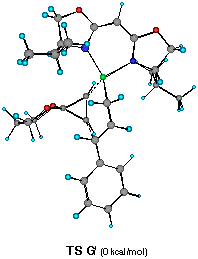
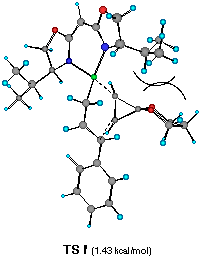
��������������������Figure 6 Top views of possible four transition structures of the addition of the chiral cinnamylzinc reagent to cyclopropenone acetal and relative heat of formation.
Each TS G', H', I' and J' gives corresponding four different cyclopropylzinc product 19, 20, 21 and 22, respectively. The energy difference corresponds to the product ratio of 85.7:8:6:0.3 at 20 oC. The enantiomeric selectivity and relative diastereoselectivity expected from the calculated difference of heat of formation are 94:6 and 92:8, respectively. Although these ratios are much higher than those obtained experimentally (i.e., 78:22, 73:27), the computational result correctly predicts the formation of 19 as a major diastereomer and the formation of the minor diastereomers 21 and 22 derived from TS H' and TS J'.
In TS G', one can observe the minimum steric interactions between the isopropyl group of ligand 2a, the phenyl substituent of the cinnamylzinc moiety, and the acetal moiety of 16. In contrast, significant steric repulsion between the acetal moiety of 16, phenyl